By Zev Kronenberg, Senior Engineer of Bioinformatics at PacBio
Since the introduction of HiFi reads the community has embraced these long and highly accurate reads for human genome assembly and paralog resolution [1-5]. At PacBio, the assembly team (Figure 1) is working to build on the accuracy of HiFi data for direct phasing during assembly.

In diploid organisms, phasing an assembly means separating the maternally and paternally inherited copies of each chromosome, known as haplotypes. Each phased contig, or haplotig, is made up of reads from the same parental chromosome (Figure 2). Phased genomes give better quality than collapsed genomes; they provide allelic information, which can be important for studying human diseases, crop improvement, evolution, and more.

Figure 2. Phased de novo assembly. A collapsed haploid assembly meshes contigs from different haplotypes (unphased assembly), while a partially phased assembly may still switch between the two haplotypes in its primary contigs. A fully phased assembly would cleanly separate the two haplotigs.
FALCON-Unzip is a diploid-aware genome assembler that has been used to assemble and phase many PacBio genomes [6]. It first creates a collapsed assembly, then uses heterozygous single nucleotide variants to partition the reads by haplotype and reassembling them into haplotigs. The assembly outputs are primary contigs with associated haplotigs (Figure 3).

Figure 3. FALCON-Unzip phasing and haplotig assembly steps. In the first stage primary contigs and associate contigs are produced, reads are aligned to the primary contigs, and phased. The phase is then re-introduced to the assembly graph, followed by re-assembly.
While FALCON-Unzip has consistently given our users excellent results, it was built for long reads with higher error rates and does not take advantage of the high accuracy of the HiFi reads. In 2019, FALCON-Unzip was adapted for HiFi data, producing high-quality results [7]. However, the current implementation still requires iterative assembly, and does not use indels for phasing. Therefore, we have started working on a new graph cleaner called Nighthawk that simplifies the assembly graph by removing cross-haplotype alignment overlaps, which can significantly speed up and improve assembly. While still a work in progress, the preliminary results are promising.
Nighthawk: A smart, efficient assembly graph cleaner
Nighthawk uses that classical bioinformatics data structure, the De Bruijn graph, to identify genetic variants (substitutions, insertions, and deletions) and remove cross-haplotype overlaps in the assembly string graph.
Most long-read genome assemblers follow the overlap-consensus-layout (OLC) workflow. The overlap stage begins with a pairwise alignment of all reads (Figure 4A). For each read, a pile of alignments to all other reads is generated. The goal of Nighthawk is to detect and remove cross-haplotype overlaps — that is, alignments between reads that come from different haplotypes. It also needs to remove other false alignments that come from paralogs, repeats, etc.
Given a pile of reads, Nighthawk builds a read-colored k-mer De Bruijn graph [8], where each node represents a k-mer; node colors denote a unique set of reads (Figure 4B). For each read overlap, Nighthawk calculates a read similarity score (RSS). The RSS is the number of shared variants between two reads. A positive RSS indicate that reads are in phase with another, while a negative RSS suggest the read overlap is cross-haplotype and should be removed (Figure 4C). Nighthawk removes overlaps with a negative RSS. The remaining overlaps are then passed on for the layout and consensus stage of assembly (Figure 4D).
It is amazing to see how clean a HiFi-based De Bruijn graph is (Figure 5). This is often a work of art in itself! After running Nighthawk, the overlaps can then be passed into string graph assemblers such as FALCON for assembly.

Figure 4. The Nighthawk workflow. Nighthawk builds a colored De Bruijn graph from read overlaps. Overlaps are scored by shared variants between two reads. Overlaps with negative RSS indicate cross-phase overlaps and are removed. The resulting overlaps are passed to a string graph assembler (such as FALCON) for phased assembly.

Figure 5. A HiFi De Bruijn graph for a pile of reads from Drosophila genome sequencing. Each dot represents a k-mer (k=23), the edges denote neighboring k-mers. The larger red dots mark the head of heterozygous bubbles.
Testing Nighthawk on a HiFi data set
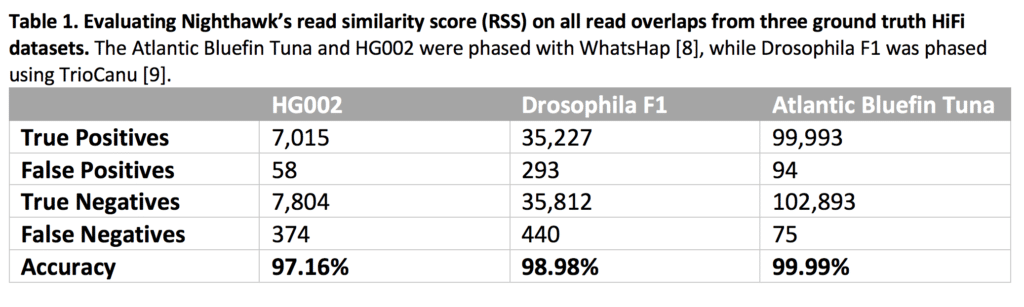
We evaluated how well Nighthawk’s RSS could distinguish in-phase and cross-phase overlaps against three ground truth sets (Table 1). In all three data sets, Nighthawk’s RSS was able to distinguish in-phase read overlaps (true positives) from cross-phase read overlaps (true negatives) while having very few false positives and false negatives.
But what effect does Nighthawk’s graph cleaning have on the assembled genome? Our team patched Nighthawk into FALCON and assembled a heterozygous (0.6%) F1 Drosophila HiFi data set. The haploid genome size is 140 Mb, so a perfectly assembled diploid genome would consist of a total of 280 Mb total in primary and associated contigs.
Our Nighthawk-FALCON assembly produced 247.1 Mb of primary contigs and 14.9 Mb associated contigs, creating a diploid genome that’s a total of 262 Mb (93.9%). The phasing accuracy, as measured by parental k-mers, was much better using Nighthawk for both primary and associated contigs compared to other methods.

Toward a truly phased assembly
We have shown that HiFi data alone can be used to effectively phase a Drosophila genome. Our new tool, Nighthawk, is an assembly graph cleaner that uses the accuracy of HiFi reads for variation detection. The phasing of the primary and associate contigs improves compared to FALCON when Nighthawk is used to filter out cross-phase alignment overlaps.
Nighthawk is still a work in progress, and many challenges remain. One such challenge is the use of alignment identity as a filter to identify cross-phase overlaps. Setting the right identity threshold is a Goldilocks problem: a filter that’s too stringent would fragment the assembly, while a filter that’s too relaxed would not remove all the false overlaps. Another challenge is complex graph structures that may arise from repeat structures, homozygosity, lack of overlap coverage, etc.
Nighthawk is only the first piece in the overlap-layout-consensus assembly process. Our team is continuing to modify string-graph algorithms to recognize the graph structures Nighthawk generates. We are excited about the new possibility HiFi data brings and believe that fast, direct phased assemblies will be feasible in the not-too-distant future.
Acknowledgments
The PacBio assembly team would like to thank Tobias Marschall (@tobiasmarschal) for the inspiration to use De Bruijn graphs for variant calling (NCBI Hackthaon 2019) and Mark Chaisson (@mjpchaisson) for technical guidance on avoiding common pitfalls.
References
[1] Wenger et al., “Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome”, Nature Biotechnology (2019)
[2] Vollger et al., “Improved Assembly and Variant Detection of a Haploid Human Genome Using Single-Molecule, High-Fidelity Long Reads”, Annals of Human Genetics (2019)
[3] Vollger et al., “Long-Read Sequence and Assembly of Segmental Duplications”, Nature Methods (2019)
[4] Garg et al., “Efficient Chromosome-Scale Haplotype-Resolved Assembly of Human Genomes”, bioRxiv (2019)
[5] Porubsky et al., “A Fully Phased Accurate Assembly of an Individual Human Genome”, bioRxiv (2019)
[6] Chin et al., “Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing”, Nature Methods (2016)
[7] Kronenberg et al., “High-quality Human Genomes Achieved through HiFi Sequence Data and FALCON-Unzip Assembly”, ASHG Poster (2019)
[8] Garg et al., “A Graph-Based Approach to Diploid Genome Assembly”, Bioinformatics (2018)
[9] Patterson et al., “WhatsHap: Haplotype Assembly for Future-Generation Sequencing Reads.” In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) (2014)
[10] Koren et al., “De Novo Assembly of Haplotype-Resolved Genomes with Trio Binning”, Nature Biotechnology (2018)
January 13, 2020 | Human genetics research