Countries around the world have recognized the value of precision health and population genomics research initiatives. They have launched efforts to sequence thousands to millions of genomes in order to learn more about how complex or rare inherited diseases impact their populations and how to mitigate their impacts. The goal? Building diverse health databases, learn how biology and lifestyle affect health, and bring us closer to a future where precision medicine is the standard.
But a population genomics program database can only be as good as the data it collects. Which raises the question – are the programs in place today able to collect everything they need to be the most effective for their target population?
Are you seeing everything?
Most population genomics programs have relied on short-read sequencing technology as the standard. However, a preprint from research groups involved in the All of Us (AOU) program in the U.S. has boldly challenged this status quo. We recently posted about a technical pilot study comparing short- and long-read sequencing that reveals HiFi long-read sequencing is a superior method for sequencing medically relevant genes. HiFi sequencing allows for the identification of more variants at lower depth than legacy methods, as well as the discovery of previously hidden variations – something that has not been achievable with current short-read technology.
A 2022 paper, “Curated variation benchmarks for challenging medically relevant autosomal genes,” reveals a significant challenge in accurately analyzing certain medically relevant genes in a clinical setting. Variant benchmark sets, provided by the Genome in a Bottle Consortium, exclude nearly 400 medically relevant genes due to their repetitiveness or polymorphic complexity. The researchers characterize 273 of the 395 challenging autosomal genes using a haplotype-resolved whole-genome assembly.
In another publication, “Characterizing the genetic polymorphisms in 370 challenging medically relevant genes using long-read sequencing data from 41 human individuals among 19 global populations,” the authors describe some genetic variants that are specific to either an individual or a continental superpopulation. These findings suggest the importance of considering genetic background in future genetic testing and drug design studies.
In the same paper, the authors conducted a side-by-side comparison of short variant calling using NGS and long-read sequencing data from the same individuals. The results revealed that long-read sequencing data has a superior capacity for resolving short variants in regions that are known to be challenging for NGS data, such as segmental duplication and low complex/simply repeat regions. Within these regions, numerous “high impact” variants were identified that could disrupt normal protein functioning. These findings shed new light on the importance of using long-read sequencing to pave the way for more accurate genetic analyses and personalized medicine.
A dark genes panel solution
That body of work, as well as additional research, inspired scientists to find solutions to some of these challenges. Dr. Fritz Sedlazeck and his team at Baylor College of Medicine’s Human Genome Sequencing Center led a project to optimize a gene panel for HiFi long-read technology. This project aimed to address the missing variant detection power in medically relevant genes and advance our understanding of genetic diversity in selected gene targets. Twist Bioscience and PacBio collaborated with the team to achieve several goals:
- Design of a probe-capture based enrichment for 389 selected genes, which are poorly covered (<90%) by current short-read platforms
- Drive towards uniform and complete coverage for genes involved in multiple diseases (see figure below)
- Develop a cost-effective and scalable solution for PopGen studies and clinical research
- Create a variant calling pipeline for SNVs, indels, SVs including haplotype phasing, along with specialized tools for some of the most challenging genes like CYP2D6 and SMN1/SMN2.
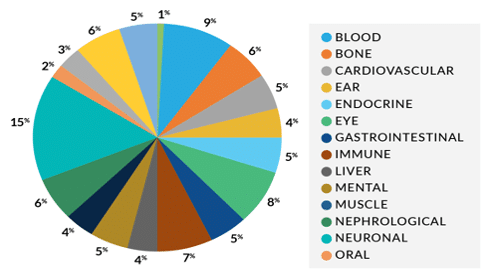
This results in low read depth and poor sequencing yield for short-read NGS and, most critically, may require additional platforms like qPCR, MLPA or microarrays to adequately capture complex variation. The Dark Genes panel will allow researchers to consolidate all the hard-to-sequence genes in a single, cost-effective, long-read targeted assay.
If you would like to discuss how your program can benefit from HiFi long-read sequencing, please ask to speak to a PacBio scientist.