Explore HiFi genome demo datasets on the Vega benchtop system
You can now bring highly accurate HiFi sequencing in-house with the power of the Vega system. HiFi long-read sequencing improves discovery of all variants including difficult-to-detect variants, structural variation and methylation changes involved in diseases and genetic diversity that are challenging or impossible to detect with short reads1.

Powerful ways scientists are using HiFi long-read sequencing
Human whole genome sequencing
Completely characterize human genomes to better understand the complexity of health and disease.
Plant + animal whole genome sequencing
Quickly and affordably produce contiguous, complete, and correct de novo assemblies of even the most complex genomes.
Microbial whole genome sequencing
Achieve closed chromosomes and plasmids from even the most repeat-dense and GC-rich genomes.
Industry-leading accuracy delivers long-read results you can trust
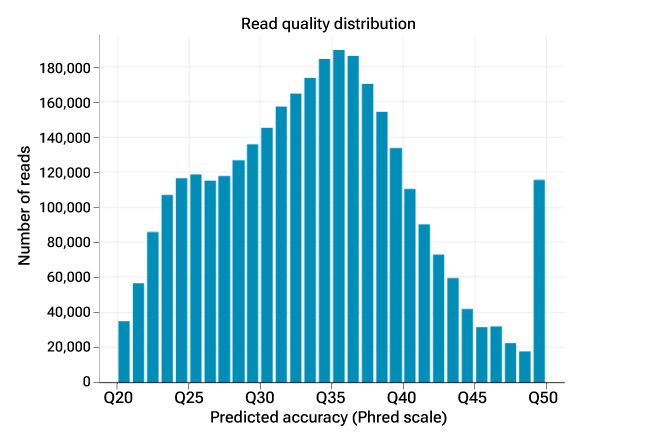
Built on proven HiFi technology and compatible with existing workflows, the Vega system brings powerful, reliable insights straight to your lab. The figure shows the read quality distribution with a median read quality of Q33 and with 90% of bases Q30 or higher. This level of accuracy is on par with standard short read technologies2 but with read lengths over 100 times longer, averaging 19.63 kb per HiFi read.

See more of the human genome with HiFi
The advent of Telomere-to-Telomere (T2T) genomes and pangenomes provides a full view of human genomic variation. Gaining that view in your own samples requires the right technology – HiFi long-reads. Explore the human HG002 (NA24385) dataset sequenced on the Vega system.
Key metrics for the demo dataset

Powerful insights: 20x HiFi long-read genome
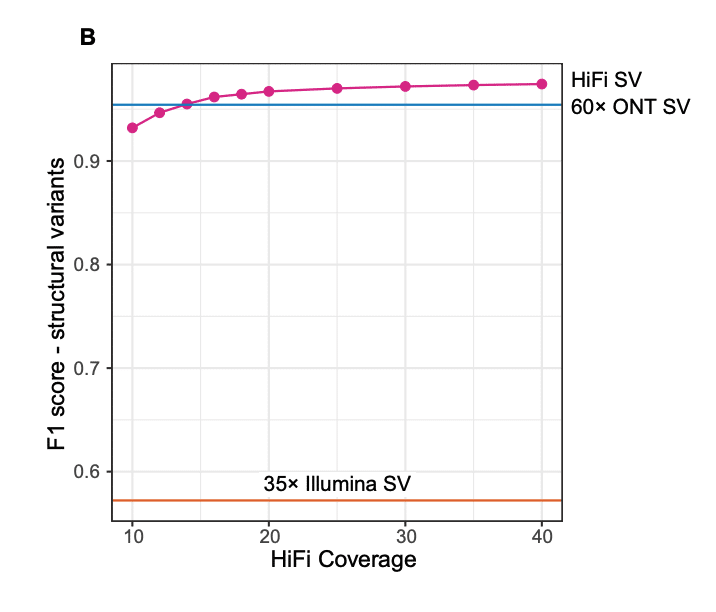
With best-in-class accuracy and long read lengths, HiFi sequencing delivers high-quality variant detection performance, even at just 20x coverage3—far surpassing other technologies like nanopore and SBS short-read sequencing, as shown in the figure. Want to explore more about what it means to generate a 20x HiFi long-read genome?
Vega shares same high-quality HiFi data as Revio
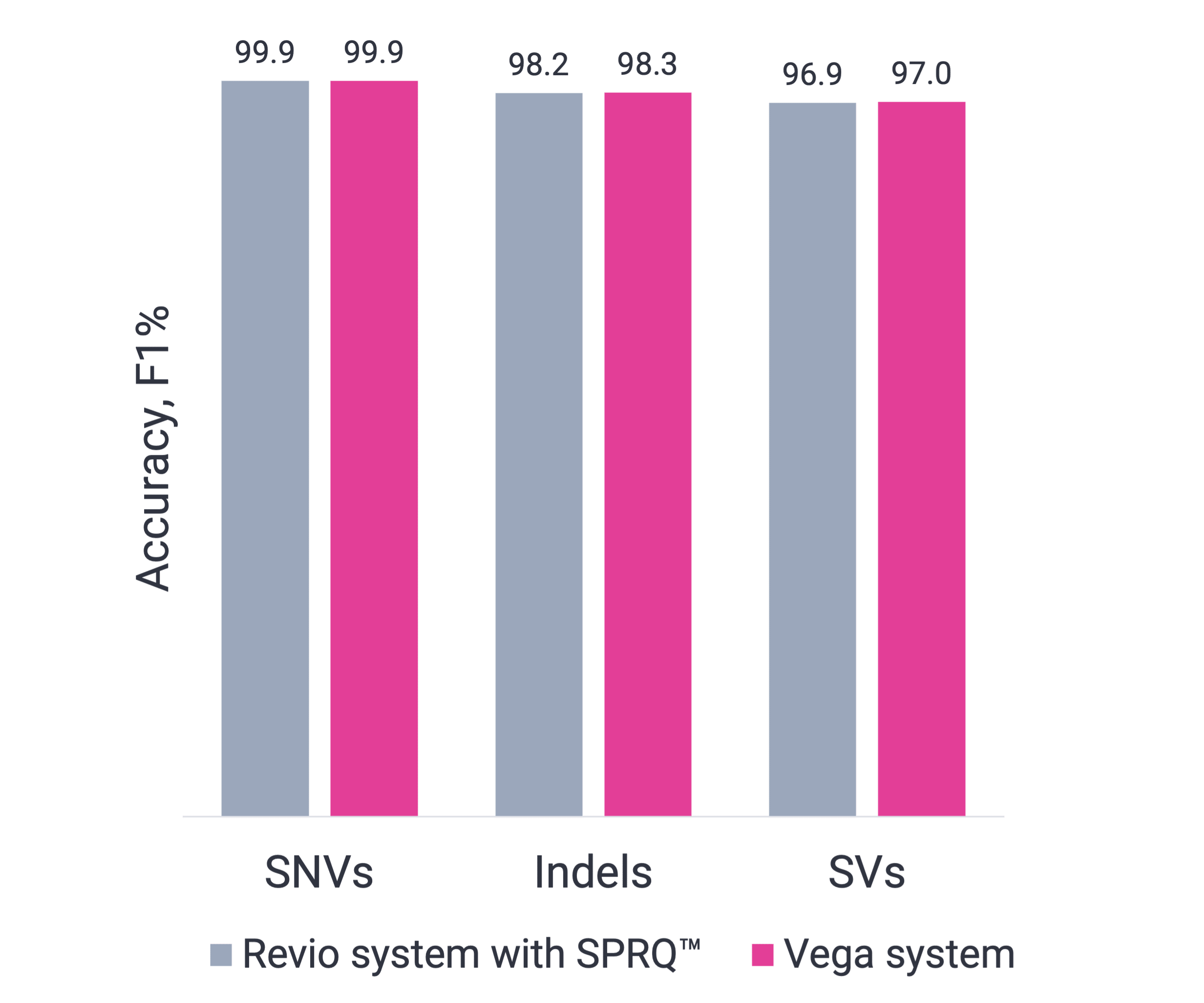
The Vega system delivers the same industry-leading accuracy as Revio but in a compact benchtop system. Shown in the figure, the HG002 human genome sequenced by the Vega and Revio systems, results in 90% of bases at Q30 or higher, with long reads that provide a full view from telomere to telomere. Whether you’re uncovering complex regions vital to disease research or detecting structural variants and repeat expansions, Vega is the essential tool you need for genomic exploration.
Plant + animal

California flannelbush

Widow rockfish

Sea otter
Explore the biodiversity of populations, species, and ecosystems
PacBio sequencing delivers data that empowers biodiversity and conservation initiatives with exceptional multiomic insights. Our HiFi long-read technology enables you to more comprehensively document and delve into the hidden wonders of the biosphere and launch campaigns to safeguard them for future generations.
Explore the California flannelbush, widow rockfish, and sea otter dataset sequenced on the Vega system and Revio v1 chemistry. Special thanks to the California Conservation Genomics Project (CCGP) & UC Davis Genome Center for providing the libraries.
Equivalent HiFi data generated from Revio and Vega
HiFi sequencing enables the most detailed reference genomes, with both Revio and Vega systems successfully assembling genomes with comparable haplotype-resolved assemblies. The same trusted HiFi data—now empowering deeper discovery and preservation of our living world.
HiFi yield and accuracy
| Revio (v1) | Vega | |||
|---|---|---|---|---|
| HiFi yield | HiFi read accuracy | HiFi yield | HiFi read accuracy | |
| California flannelbush | 65.9 Gb | 99.90% (Q30) | 64.9 Gb | 99.97% (Q35) |
| Widow rockfish | 87.1 Gb | 99.95% (Q33) | 64.9 Gb | 99.96% (Q34) |
| Sea otter | 77.0 Gb | 99.87% (Q29) | 64.9 Gb | 99.96% (Q34) |
Haplotypes 1
| Haplotypes 1 | ||||
|---|---|---|---|---|
| Sequencing system | Assembly size | N contigs | Contig N50 | |
| California flannelbush | Revio | 1.20 Gb | 211 | 25.3 Mb |
| Vega | 1.21 Gb | 217 | 25.4 Mb | |
| Widow rockfish | Revio | 820 Mb | 216 | 21.2 Mb |
| Vega | 817 Mb | 230 | 19.8 Mb | |
| Sea otter | Revio | 2.44 Gb | 685 | 64.5 Mb |
| Vega | 2.46 Gb | 538 | 55.6 Mb | |
Haplotypes 2
| Haplotype 2 | ||||
|---|---|---|---|---|
| Sequencing system | Assembly size | N contigs | Contig N50 | |
| California flannelbush | Revio | 1.24 Gb | 286 | 23.6 Mb |
| Vega | 1.24 Gb | 264 | 25.5 Mb | |
| Widow rockfish | Revio | 819 Mb | 298 | 19.2 Mb |
| Vega | 823 Mb | 269 | 22.4 Mb | |
| Sea otter | Revio | 2.46 Gb | 655 | 68.1 Mb |
| Vega | 2.48 Gb | 577 | 60.3 Mb | |
What can you expect with one sequencing run on the Vega system?
| Whole genome sequencing | Samples per Vega SMRT Cell |
|---|---|
| Human genome at 20x | 1 |
| De novo assembly (2 Gb genome) | 1 |
| Variant detection (2 Gb genome) |
Structural variants: 3 |
| Microbial genomes (total 2 Gb) | 384 |
| All sample throughputs are estimates for either the Vega system with 1 SMRT Cell. Coverage may vary based on sample quality, library quality, and fragment lengths. Microbial de novo assembly assumes microbes with 2 Gb of total genome size at 30x per sample. | |
HiFi genome workflow

Sample prep
DNA extraction with Nanobind PanDNA kit (2 hrs)
Short fragment depletion with short read eliminator kits (SRE), if necessary (2.5 hrs)
DNA shearing to 15 – 20 kb length. (8 to 30 min)


Sequencing
Vega system sequencing (24 hours)
On-instrument primary analysis:
- HiFi base calling
- 5mCpG calling
- Demultiplexing secondary analysis

Analysis
Secondary analysis using on-premise or cloud services:
- SMRT Link analysis tools and data utilities
- 5mCpG calling
- PacBio WGS Variant Pipeline (human)
References
- Höps W., et al. (2024). HiFi long-read genomes for difficult-to-detect clinically relevant variants. medRxiv, 2024-09.
- https://www.illumina.com/systems/sequencing-platforms/novaseq-x-plus/specifications.html
- Kolesnikov A., et al. (2024) Local read haplotagging enables accurate long-read small variant calling. Nature Communications,15, 5907.
- HG002 T2T draft genome benchmark: https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/NIST_HG002_DraftBenchmark_defrabbV0.015-20240215/
- Saunders, C., et al. (2024) Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling. bioRxiv, https://doi.org/10.1101/2024.08.19.608674
- Oxford Nanopore Open Data. https://labs.epi2me.io/giab-2023.05/ Accessed September 2024.
- Behera, S., et al. (2024). Comprehensive and accurate genome analysis at scale using DRAGEN accelerated algorithms. bioRxiv.
- Wagner, J., et al. (2022). Benchmarking challenging small variants with linked and long reads. Cell Genomics, 2(5).
- https://github.com/PacificBiosciences/HiFi-human-WGS-WDL
- Illumina Hap.py framework: https://github.com/Illumina/hap.py