Directly detect epigenetic modifications with HiFi sequencing
HiFi sequencing directly detects DNA modifications in native DNA from variations in the polymerase kinetics of DNA base incorporation, without the need for arduous chemical conversion protocols like bisulfite treatment.
- Detect genome-wide m6A and m4C R-M system motifs at coverage levels recommended for assembly
- Obtain complete genomes with annotations for epigenetic modification
- Reveal phase variation of R-M genes that regulate batteries of genes involved in pathogenesis, host adaption, and antibiotic resistance
Learn how other scientists have used SMRT sequencing to connect prokaryotic methylation to new biology:
- PacBio-powered analysis indicates methylation makes TB pathogen so wily
- SMRT sequencing enables discovery of epigenetic driver of C. diff persistence
- Creating an epigenetic barcode to accurately characterize microbial communities
Workflow: from DNA to microbial methylome

Sample + library preparation
- Start with unamplified genomic DNA
- Prepare SMRTbell libraries following the protocol for microbial whole genome sequencing.

Sequencing

Data analysis
- Use SMRT Link Microbial Genome Analysis to assemble and annotate active R-M system motifs
- View detailed reports on modifications and associated motifs
Spotlight
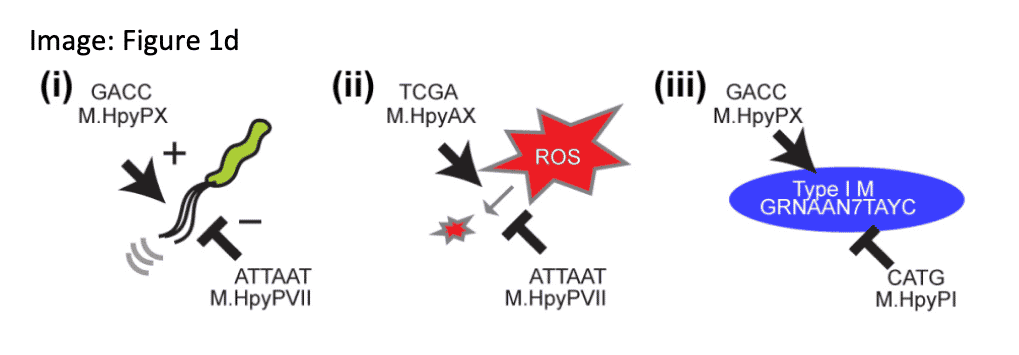
METHYLTRANSFERASE NETWORKS ENABLE NIMBLE ADAPTION IN H. PYLORI
H. pylori is known for its large and variable repertoire of active R-M systems. A series of single-gene knock-out experiments reveals these methyltransferases form a hierarchical, combinatorial, and highly adaptable gene-regulatory network that influences motility, oxidative stress tolerance, virulence, DNA damage repair, and other critical pathways related to adaptive evolution.
Yano, H., et. al. (2020) Networking and specificity-changing DNA methyltransferases in Heliobacter pylori. Frontiers in Microbiology. 11, doi: 10.3389/fmicb.2020.01628.
Spotlight
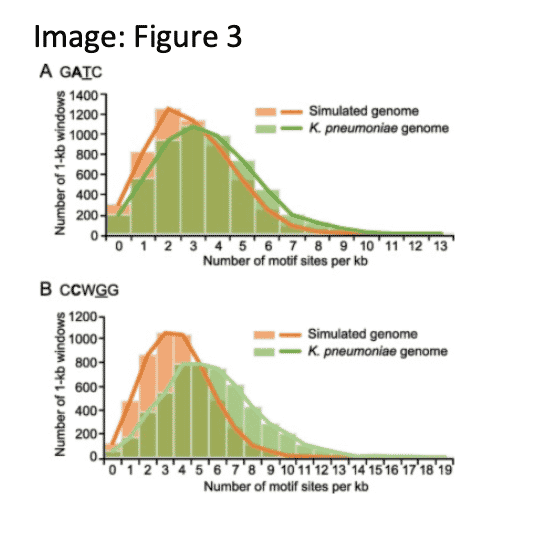
A role for epigenetics in the regulation of replication and transcription
Comprehensive characterization and comparison of the methylomes of 14 K. pneumoniae strains revealed differential concentration of GATC motifs and methylation penetrance in intergenic regions. Modelling of MTase-catalysed methylation suggests slower rates of motifs in replication origins and IGRs, implicating a role in the initiation of replication and transcription.
Fu, J., et. al. (2022). Precision Methylome and In Vivo Methylation Kinetics Characterization of Klebsiella pneumoniae. Genomics, proteomics & bioinformatics, 20(2), 418–434. https://doi.org/10.1016/j.gpb.2021.04.002
{kind=link}
{kind=link}