
The books of life are fascinating yet challenging reads, owing to the complexity of the underlying biological processes and their alterations that lead to disease. Thanks to highly accurate HiFi long-read sequencing, we are now seeing fundamental changes in our ability to print, read and comprehend this most exciting literature.
This is perhaps no more striking than in the field of studying the myriad of mRNA transcript isoforms that are expressed from a genome. The limitations of short-read RNA sequencing to resolve this complexity have long been recognized – for example, as illustrated in a perspective in Nature Methods ten years ago:
In the same issue, Hagen Tilgner and Mike Snyder (Stanford University) commented that “the way we do RNA-seq now is… you take the transcriptome, you blow it up into pieces and then you try to figure out how they all go back together again. If you think about it, it’s kind of a crazy way to do things”. They were also among the first to demonstrate a better way: full-length RNA sequencing, using PacBio’s Iso-Seq method, for “A single-molecule long-read survey of the human transcriptome”, resolving RNA transcript molecules by sequencing them in their entirety.
In a recent Nature Communications paper entitled Isoform-resolved transcriptome of the human preimplantation embryo, researchers from the Icahn School of Medicine at Mt. Sinai, Tel-Aviv University, the University of Louisville, and UC Irvine describe the first isoform-resolved transcriptome of early human development. Performing Iso-Seq (and short-read RNA-seq) on 73 embryos spanning the zygote to blastocyst stages, they “identified 110,212 unannotated isoforms transcribed from known genes… 17,964 isoforms from 5,239 unannotated genes, which are largely non-coding, primate-specific, and highly associated with transposable elements”.
Demonstrating the utility of an “isoform-resolved approach to comprehensively profile RNA expression and splicing during these critical stages of development”, they report that “alternative splicing and gene co-expression network analyses further reveal that embryonic genome activation is associated with splicing disruption and transient upregulation of gene modules”, and concluding that “these findings show that the human embryo transcriptome is far more complex than currently known and will act as a valuable resource to empower future studies exploring development.”
As a second example, in a new preprint entitled Characterization of Alternative Splicing During Mammalian Brain Development Reveals the Magnitude of Isoform Diversity and its Effects on Protein Conformational Changes, researchers from Dresden, Germany and Lausanne, Switzerland, present a “new avenue for assessing gene function in cell fate commitment by looking at the potential diversification of protein structure resulting from AS [alternative splicing] rather than, as universally adopted, measuring gene expression alone”.
The researchers leverage Iso-Seq to “reconstruct cell type-specific transcriptome diversity during brain development and quantitatively assess AS events”, describing “nearly 50,000 new transcripts including novel exons, splice sites and/or microexons, thus, uncovering the full spectrum of splicing dynamics accompanying [cell] fate transitions.”
Excitingly, the authors go one step further and “computationally infer the biological significance of AS on protein structure by using AlphaFold2”, finding that “nearly 40% of isoform pairs originating from the same gene exhibited large global conformational changes including fold switches”. They describe the widespread “occurrence of regions with identical sequence yet adopting profoundly different secondary structures …, depending on distant AS events, thus, revealing that even negligible changes in exon usage can induce large conformational changes influencing the functional properties of proteins.”
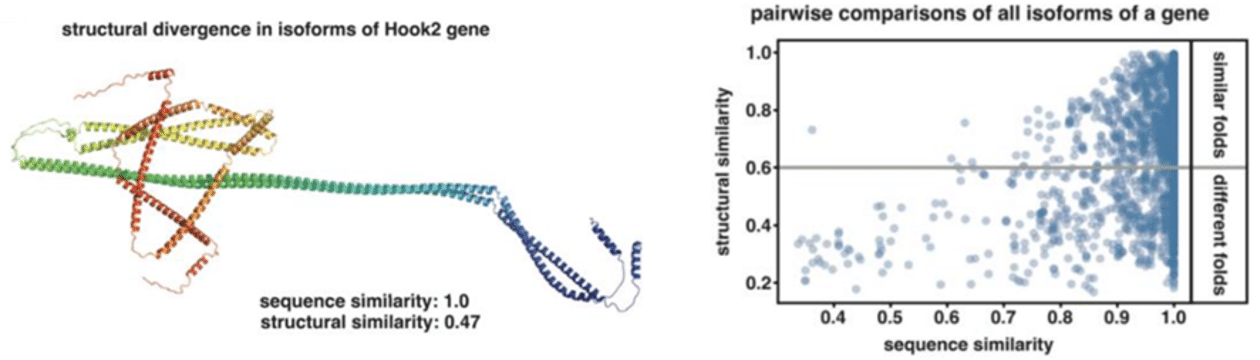
The preprint’s conclusion that “AS has a greater potential to impact protein diversity and function than previously thought independently from changes in gene expression” supports an ever-increasing recognition that transcript isoforms, not genes, determine biology and disease.
While powerful, until now Iso-Seq studies run on Sequel IIe systems have been limited due to their relatively low throughput of generating full-length transcript reads. In the above analogy, perhaps this was akin to writing books with a typewriter. Now, the combination of the Revio system and our new high-throughput Kinnex RNA sequencing kits represent the transition to a ‘printing press’ for high-quality RNA sequencing, allowing orders of magnitude higher throughput to generate comprehensive, full-length transcriptome data in a cost-effective and efficient workflow. We cannot wait to see what discoveries you will make by reading your research books of life in this new way, from cover to cover. Please connect with us to let us assist you in your next RNA sequencing project.