In living cells, it is typically the proteins that are the work horses of carrying out the myriad of biological functions, and that can be subject to aberrant processes that cause disease. With a limited number of genes, the incredible spectrum of different protein functions is brought about by protein isoforms, commonly interrogated with peptide mass spectroscopy, that result from differential splicing of mRNAs from the same gene. In a new review article by Dr. Gloria Sheynkman (University of Virginia) and collaborators Dr. Pete Castaldi and Arby Abood, entitled Bridging the splicing gap in human genetics with long-read RNA sequencing: finding the protein isoform drivers of disease, the incipient opportunities and benefits of full-length RNA sequencing for the field of proteogenomics are highlighted.
The authors describe that many human diseases are caused by aberrant splicing, including cancer, cardiovascular diseases, and neurological disorders. The concept of underlying genetic splicing quantitative trait loci (sQTL) is reviewed, stressing that the identification of the corresponding isoform or protein products associated with disease-associated sQTLs is challenging with short-read RNA-sequencing methods, noting that they “cannot precisely characterize full-length transcript isoforms”. In addition, many reference transcript annotations are still based on short-read RNA-seq, which are thereby incomplete. The paper highlights how long-read sequencing offers solutions to these shortcomings by sequencing full-length mRNA transcripts and linking sQTLs to full transcript isoforms containing disease-relevant protein alterations, directly linking RNA isoforms to protein-level functions.
As one specific example, Dr. Sheynkman & colleagues applied PacBio’s Iso-Seq method of highly accurate, full-length RNA sequencing to endothelial cells in a recent preprint. Full-length transcripts delineated from PacBio sequencing served as the basis for a protein database used for downstream mass spectroscopy peptide analysis to infer protein isoform expression.
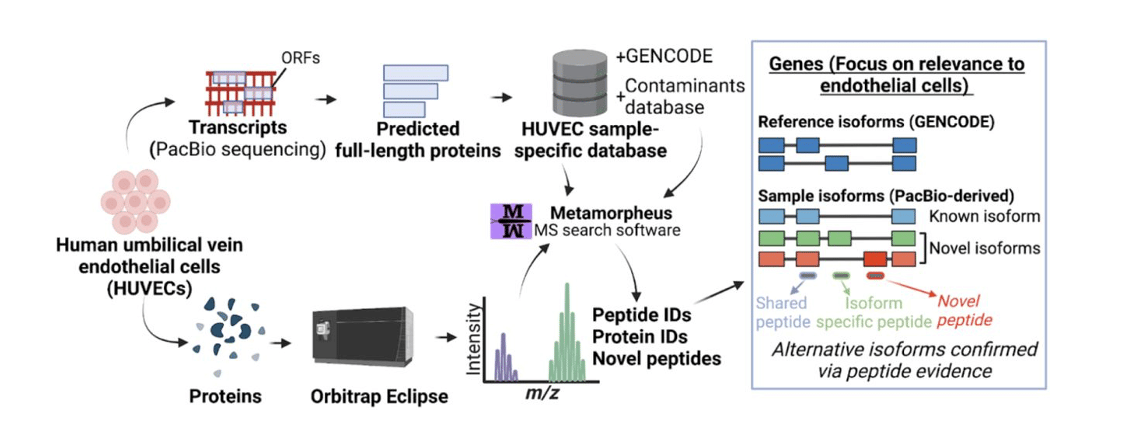
The researchers detected over 50,000 transcript isoforms from over 10,000 genes – remarkably, over 22,000 of these transcript isoforms were completely novel, having never been observed before. Further, about a quarter of the time, the predominant isoform “does not correspond with the accepted ‘reference isoform’”. And upon incorporating Iso-Seq’s full-length transcript evidence, the researchers nearly doubled the number of detected protein isoforms, including important endothelial-related genes and associated signaling pathways. Together with the researchers’ first publication on using PacBio’s Iso-Seq method for enhancing proteogenomics, noting that “the PacBio-derived proteome differs substantially from the reference proteome”, the researchers conclude:
Among long-read sequencing methods, PacBio’s highly accurate, full-length Iso-Seq method has been demonstrated as the best-in-class technology, with numerous significant advantages and benefits, including:
- PacBio’s Iso-Seq method captures longer transcripts (e.g., average 2.1 kb (Miller et al.) or 3.3 kb (Tseng et al.)) than other methods
- PacBio’s Iso-Seq method better captures the 5’ and 3’ ends (e.g., 50-80% (Veiga et al.) or 50-70% (Tseng et al.), meaning that new isoforms with novel start and end sites can be discovered without relying on aggressive filtering for known annotated sites
- PacBio’s Iso-Seq method produces highly accurate transcripts that can be used directly for ORF prediction, recovering more peptides in mass spectrometry data (e.g., 99% of peptides using a PacBio + GENCODE database (Miller et al.))
- PacBio’s Iso-Seq method better resolves allele-specific isoform expression (Wang et al., Tian et al.), and with fewer reads required to accurately call small variants (de Souza et al.)
As described in a recent community perspective on the roadmap for the functional annotation of protein families, it is estimated that currently only half the predicted sequenced proteins carry an accurate functional annotation. With only one (1) SMRT Cell 8M required to characterize a whole human transcriptome, PacBio HiFi sequencing constitutes a powerful new method to aid in our understanding of the role of the proteome in health and disease, with many profound discoveries just around the corner.
Please join us for our Iso-Seq Social Club vol. 3 event on September 14, which among other speakers will feature Madison Mehlferber from Dr. Sheynkman’s group to describe this transformative proteogenomics research in more detail.