Get more from your HiFi reads with HiFiCNV, a powerful new software tool for capturing large copy number variants throughout the genome.
Why CNVs and HiFi reads are important
Copy number variants (CNVs) are large sequence gains or losses induced by structural variants. Because their size and ability to impact multiple genes make them a common source of genetic diseases, CNVs are a prime target for researchers focused on identifying putative sources of illness within an individual’s genome.
PacBio HiFi reads are exceptionally useful for detecting structural variants because they are both highly accurate (90% of bases ≥ Q30) and long (~15 to 20 kb on average), with increased mappability over traditional NGS data. However, at present most tools designed to detect CNVs have been optimized for shorter reads.
An innovative tool for CNV discovery
To unlock the full potential of HiFi reads for identifying large CNVs across the genome, we developed HiFiCNV. When used in conjunction with the expanding suite of PacBio genomics tools, this powerful new software solution will enable users to identify all major classes of genetic variation with PacBio HiFi reads.
To evaluate CNV detection accuracy, we compared results from HiFiCNV to a collection of known CNVs described in a study by Gross et al. This study included 17 publicly available samples with clinically relevant events spanning sizes from 10 kb to full chromosome duplications. HiFiCNV detected all large CNVs from this dataset, and 90% of those calls had high overlap accuracy (>70% overlap) when compared to the reported CNV.
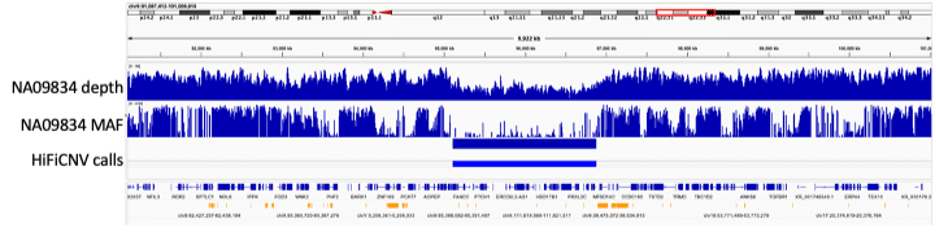
In addition to CNV detection, HiFiCNV can generate several CNV related track files which can be loaded into IGV for visualization and assessment of its variant calls. The figure below illustrates the use of these tracks for sample NA09834. This sample contains a copy number loss spanning PTCH1, a gene associated with the sample’s phenotype, basal cell nevus syndrome. The figure shows the copy number loss region along with the CNV call, read depth tracks, and minor allele frequency tracks generated by HiFiCNV. For this large event, spanning ~1.7 Mbp on chr9, the associated depth and allele frequency tracks help to visually illustrate evidence for the called copy number loss.
With HiFiCNV, large copy number variants can be conveniently identified from HiFi data and visually reviewed when needed. When combined with the variation generated from other HiFi tools, researchers can access a more complete view of all types of variation present in their samples, enabling more accurate analysis of rare diseases.