 Microbial and metagenomics enthusiasts rejoice: newly optimized bioinformatics pipelines are enabling scientists to detect even more species and assemble more single contig metagenomes using HiFi sequencing than ever before – with up to 9-fold increases in some cases.
Microbial and metagenomics enthusiasts rejoice: newly optimized bioinformatics pipelines are enabling scientists to detect even more species and assemble more single contig metagenomes using HiFi sequencing than ever before – with up to 9-fold increases in some cases.
Due to the continuous advances, improved results, and cost-effectiveness of PacBio HiFi long-read sequencing, it is an increasingly popular tool for metagenomic analysis of complex microbial communities.
Recently, the PacBio team has been working to update our tools and pipelines to ensure that they are fully optimized for HiFi data and can provide even more value to metagenomic studies.
The result? Metagenome taxonomic profiles that yield more classified and low abundance species with higher precision, and shotgun metagenome assemblies that return more high-quality and complete, circular, single contig metagenome assembled genomes (MAGs).
Digging into the data
To test the effectiveness of the new pipelines, we worked with pooled human gut microbiome samples from the BioCollective.
Two samples came from vegan donors and two from omnivore donors, allowing us to see how diet influences gut microbiota. The pooling process, which creates a reference material by pooling samples from multiple donors (in this case four adults), leads to a more complex sample and a richer data set than can be obtained through mock community approaches. It also gives a more consistent composition than samples from an individual donation. As a bonus, the dataset is available for anyone to work with.
Each sample was sequenced on a SMRT Cell 8M using the Sequel II system, resulting in nearly 2 million reads per sample, with mean read length close to 10 kb and median qualities of Q40.
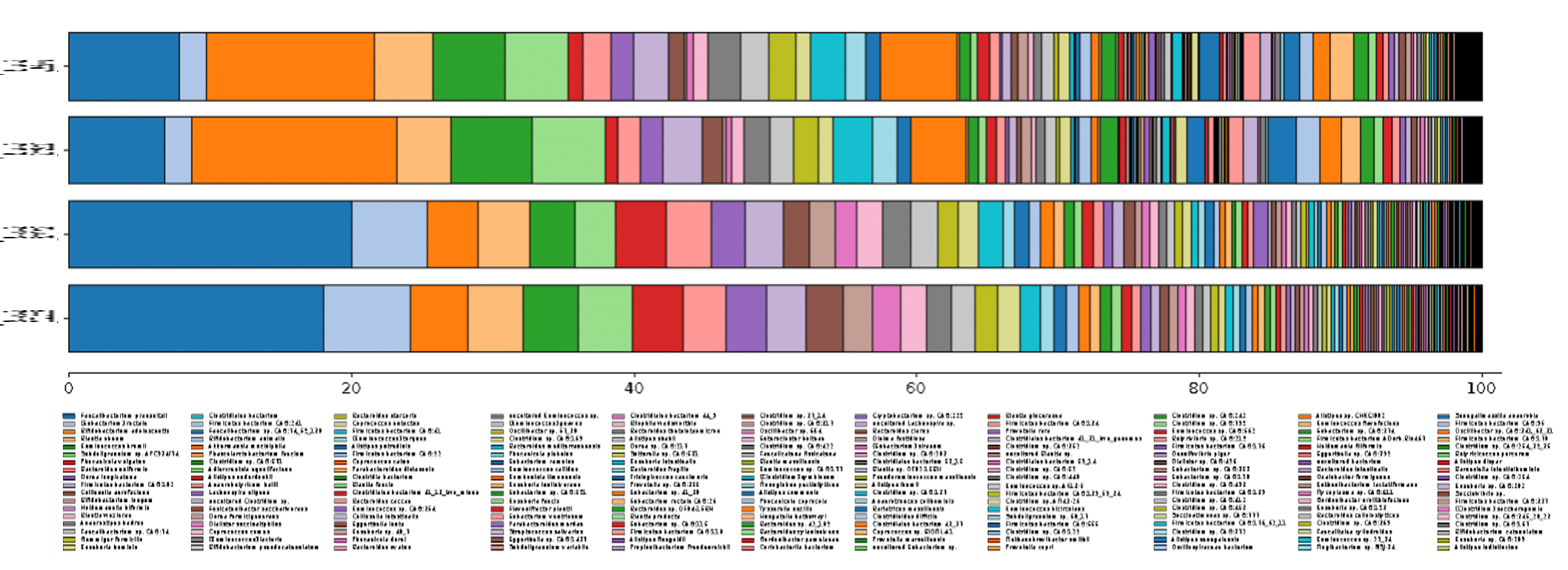
Using the updated bioinformatics pipelines, we conducted shotgun metagenome taxonomic profiling, and were able to detect 199 species at the optimized detection setting, and 690 species at the ultra-sensitive setting (Figure 1).
Compared to previous profiling efforts which detected 76 species at conservative detection settings, that is an increase of 123-614 species classified from the same data – a 162% – 808% rise!
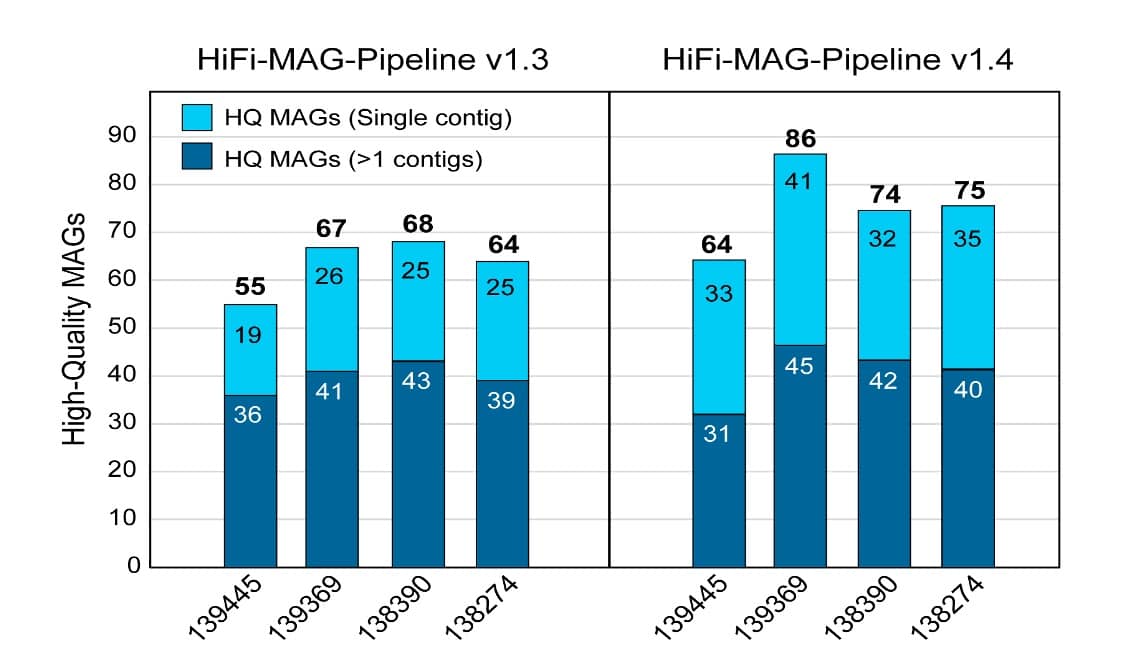
Next, we undertook shotgun metagenome assembly, aiming for high quality contig MAGs that were at least 70% complete, with less than 10% contamination, and under 10 contigs. While previous efforts produced 55-70 high-quality MAGs per sample, 25 of which were contained within single contigs, and about 126 species/strains, the updated bioinformatics pipeline combined with a new circular-aware binning strategy produced 65-85 high quality MAGs per sample, with 35 single contigs and about 143 species/strains (Figure 2).
That is an increase of 45 high quality MAGs and 46 single contig MAGs from the same data, or increases of 18% and 48%, respectively.
We are excited that these improvements will enable our customers to get even more out of samples’ data, and out of each SMRT Cell. Truly high-quality MAGs, and single-contig genomes with no assembly required allow for greater insights and accelerated scientific discovery – all things that are definitely worth celebrating.
To learn more about how you can integrate HiFi into your metagenomic workflows, visit our dedicated Microbiology and complex populations pages, as well as an overview of HiFi sequencing.
Additional resources:
● PIPELINE: PacBio metagenomic bioinformatic tools
● DATASET: BioCollective pooled gut microbiome samples
● BLOG: Data release: human microbiome samples demonstrate advances in HiFi-enabled metagenomic sequencing