In what one reviewer described as “a landmark paper for genomics,” the Cell study describes 26 PacBio-based de novo assemblies selected from 2,898 globally collected soybean germplasms for their phylogenetic relationships and geographic distributions.
Soybeans have an astounding 60,000 accessions adapted to different ecoregions, and intergenomic comparisons have shown extensive genetic diversities between wild and cultivated soybeans, as well as among cultivated soybeans from different geographic areas.

Two other reference genomes were released in 2018 and 2019, one from ‘‘Zhonghuang 13’’ (ZH13), the most widely planted soybean cultivar in China, and another from a wild soybean. Comparisons between Wm82 and these two genomes further demonstrated that a considerable amount of CNVs and PAVs exist in different accessions.
Through a comparative genome analysis of the 26 genomes, plus three previously reported genomes, Zhixi’s team identified a total of 14,604,953 SNPs and 12,716,823 small insertions and deletions; 723,862 present and absent variations; 27,531 copy number variations; 21,886 translocation events; and 3,120 inversion events.
“The high-quality genomes enabled the identification of numerous complex variations that cannot be detected by simply mapping the short reads to a single genome,” the authors wrote.
They said de novo construction of the pangenome using long-read technology was valuable in identifying larger structural variants; most of the PAVs had a length from 1 kb to 2 kb, translocations were concentrated from 10 kb to 30 kb, inversions mainly ranged from 100 kb to 200 kb, and the CNVs varied from 2 to >10 with an enrichment of 2 and 3.
“The large number of SVs from dozens of independently de novo assembled genomes enabled us to clarify clearer evolutionary processes that cannot be detected from one or a few genomes,” the authors wrote.
This will be key in the creation of new experimental populations for both functional studies and breeding, especially as genetic diversity bottlenecks during soybean domestication and improvement has resulted in narrowed genetic diversity among modern cultivars of the important crop, they added.
In order to overcome some of the limits of conventional linear references, which are unable to show the genotypes of different alleles from each locus, the team devised a way to represent it graphically, creating the first reported graph-based plant genome.
“The graph-based genome offers a new platform to map short-read data to determine the genetic variations at the pan-genome level instead of a single genome and prevent erroneous variation calls around SVs,” they wrote.
Coupled with RNA sequencing data from individual accessions, the platform will make it possible to link SVs with gene expression, which could greatly promote gene discovery. The graph-based genome could also provide an opportunity to re-analyze and “rejuvenate” previous sequencing data to generate more comprehensive information than ever, they said.
This pangenome collection, along with the methods of generating a graph-based representation of the soybean pangenome, will serve the genomics community greatly as we embark on larger projects to characterize all variation within a species for better breeding programs and conservation efforts.
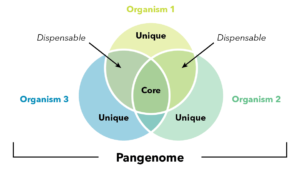
 Graphical abstract of the new 26-line soybean pangenome.
Graphical abstract of the new 26-line soybean pangenome.To learn more about pangenomes, watch the webinar Beyond a Single Reference Genome – The Advantages of Sequencing Multiple Individuals.
See additional examples of the use of SMRT Sequencing for the generation of pangenomes:
- Project to Rapidly Sequence Maize Pangenome Delivers Publicly Available Resource
- Sequencing 101: Looking Beyond the Single Reference Genome to a Pangenome for Every Species