
A few weeks ago, we described the use of HiFi sequencing to drive the paradigm shift from a single reference genome to a pangenome derived from a diverse set of individuals, using the alga Chlamydomonas reinhardtii as an example. This shift has now arrived for the human genome, as described in several new preprints from the Human Pangenome Reference Consortium (HPRC) and its 52 collaborating institutions, highlighting the substantial benefits that a human pangenome provides over the current linear GRCh38 reference.
The main preprint, A Draft Human Pangenome Reference, describes a high-quality human pangenome reference built from diploid, phased, de novo PacBio HiFi assemblies of 47 genetically diverse individuals (94 haplotypes). The authors report excellent sequence completeness (>99%), gene completeness (>99%), structural accuracy (>99%), and base pair accuracy (>99.999%) for the assemblies. To build a combined representation of human genetic variation, the assemblies were combined into a pangenome variation graph. In addition to capturing known variants and haplotypes, the pangenome reveals novel alleles at structurally complex loci and adds 119 million bases (90 Mb from structural variants) of euchromatic polymorphic sequence, and 1,529 gene duplications relative to the GRCh38 reference. In addition, the encompassing 94 sets of Ensembl gene annotations represent the largest collection to date of de novo assembled human transcriptome annotations.
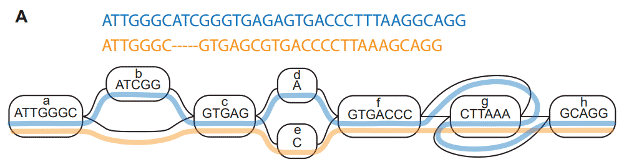
The authors describe numerous immediate benefits of utilizing the pangenome through the removal of biases inherent with the use of a single, linear reference genome:
- Improved variant calling: 34% fewer errors in small variant discovery, and 104% more detected structural variants per haplotype
- Resolution and allele frequency estimation of complex regions, including known medically-relevant loci, with the largest absolute increases in accuracy found in the Genome-in-a-Bottle (GIAB) challenging medically relevant genes benchmark
- Improved representation of tandem repeats
- Improved RNA-seq mapping
- Improved ChIP-seq analysis
The choice of using PacBio HiFi sequencing and the associated hifiasm-trio assembler for this first high-quality pangenome is described in a companion preprint. Automated assembly of high-quality diploid human reference genomes performed a detailed, comprehensive comparison of sequencing technologies and assembly software for generating the most accurate diploid genomes, concluding that HiFi-based assemblies gave the best results:
“Approaches that used highly accurate long reads and parent-child data to sort haplotypes during assembly outperformed those that did not.”
The authors report that trio-based assemblies that used HiFi reads exhibited:
- The largest haplotype phase blocks, and highest number of total phased bp
- The lowest haplotype switch errors
- The most complete separation of parental haplotypes, and least collapsed sequence
- The most accurate variant calls across single-nucleotide variants (SNVs), small indels, and structural variants (SVs) in challenging regions, exceeding existing variant benchmarks, and allowing an evaluation of haplotype differences truly genome-wide (even including regions such as centromeres)
- The highest assembly base accuracy, with less than one base call error per 100,000 bp (QV ≥ 50), which the authors directly attribute to the high accuracy of the underlying HiFi sequence data:
“Obtaining such a high degree of accuracy (QV ≥50) with long reads has only been a recent advance, due to the high base calling accuracy of HiFi reads.”
Further, as the main preprint highlights, “highly accurate haplotype-resolved assemblies allow us to access previously inaccessible regions highlighting novel forms of genetic variation … and providing new insights into mutational processes such as interlocus gene conversion.” A third preprint, Increased mutation rate and interlocus gene conversion within human segmental duplications, examines this in detail, explaining that SNVs within segmental duplications (SDs) have not been systematically assessed because of the difficulty in mapping short-read sequence data to virtually identical repetitive sequences, and “as a result … became blacklisted from subsequent genomic analyses”. Using the PacBio HiFi-based assemblies, the authors extend variant calling into 120 Mb of additional SD sequence per genome, and identify “over 1.99 million non-redundant SNVs in a gene-rich portion of the genome previously considered largely inaccessible.” Remarkably, they find “that human SNVs are elevated 60% in SDs compared to unique regions.” In other words, these previously inaccessible regions are especially rich in genetic variation across the human population, underscoring their significance.
A fourth companion preprint, Gaps and complex structurally variant loci in phased genome assemblies, perhaps most strikingly articulates the new standard of what it means to sequence a human genome:
“We no longer consider collapsed 3 Gbp genome assemblies as state-of-the-art (i.e., one representation of an individual where both haplotypes are merged) but instead consider two genomes for every diploid genome assembled (i.e., 6 Gbp vs. 3 Gbp) where parental haplotypes are phased and fully resolved.”
The authors “find that trio-based approaches using HiFi are the current gold standard” for producing such assemblies. They characterize a few challenges that must be resolved for these 6 Gb genomes to achieve the full telomere-to-telomere contiguity reached for the 3 Gb genome of the haploid CHM13 cell line. The authors propose advances in automated assembly algorithms, understanding of human genetic variation, and DNA sequencing technologies to achieve this goal.
These new preprints offer a powerful picture of how complete, accurate, and contiguous human genomes from HiFi reads advance the entire field of human genomics research. We are honored to work with the scientific community to help drive these advances and look forward to connecting with researchers across the globe to leverage many additional HiFi-based reference genomes and population genomics datasets to further improve our understanding of human biology and better human health.
Contact us to discuss how you can produce reference pangenomes for your studies as well.
Read more about the HiFi Difference
Enabling the era of reference pangenomes
More precise genomes for precision medicine
Not all gigabases are created equal
Haplotype phasing in genome assembly
True long reads vs. synthetic long reads
For more information on plant and animal genomics, go here.