Through the development of long-read, full-length RNA sequencing methods, the scientific community has recognized a fundamental paradigm shift, “from ‘gene-centric’ to ‘isoform-centric’ understanding of human transcriptomes, owing to the frequent generation of diverse transcripts from a single genomic locus” (Jing et al. (2019) Oncogene 38: 3047). PacBio’s Iso-Seq implementation has been employed in numerous publications, allowing for new insights into transcriptome complexities and their implications for understanding biology and disease. Unlike short-read RNA-seq, which observes small fragments of transcripts, long-read, full-length RNA sequencing directly observes entire spliced isoforms.
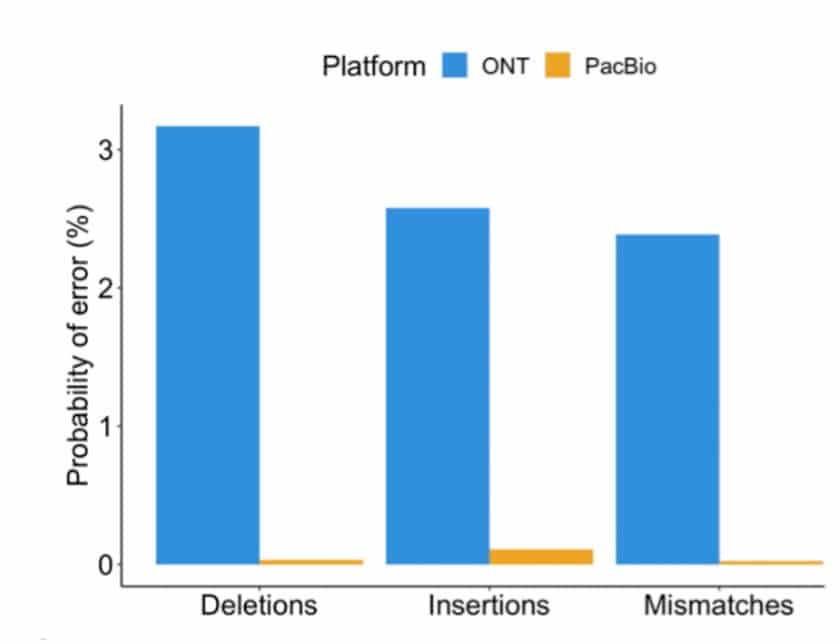
A detailed comparison of long-read technologies for full-length RNA sequencing was recently described in a new publication by the lab of Hagen Tilgner at Weill Cornell Medicine and collaborators, using a clever barcoding strategy to sequence cDNA copies from the same RNA molecule with the different sequencing technologies. As described in the paper, the PacBio HiFi Iso-Seq reads were significantly more accurate (see figure below). The lower-accuracy ONT reads led to sequence-error induced inaccurate ONT alignments, including missing alignments of short exons. PacBio’s higher read accuracy also translated to a much higher barcode recovery, 85% for PacBio vs. 16% for ONT. Notably, most of the PacBio transcript reads were also longer than ONT, and more often contained elements important for defining complete isoforms. The authors also observed that PacBio reads performed better for identifying transcript start sites, splice junctions and the mapping of short exons.
When it comes to short-read RNA-seq, a new preprint from researchers at OHSU evaluated the calling of intron retention transcriptional events from short-read data using PacBio’s Iso-Seq data as the baseline of truth. It turned out that event calling from short-read RNA-seq was not adequate, and the authors concluded “We find that short-read tools detect intron retention with poor recall and even worse precision, calling into question the completeness and validity of a large percentage of putatively retained introns called by commonly used methods.”
PacBio’s Iso-Seq method provides the highest quality for cDNA sequencing, providing confidence of the underlying data that illuminates composition, dynamics, and disease-implicated aberrations of transcriptomes. In a recent example, this paper shows how Dr. Jacques Banchereau (Jackson Laboratory) & collaborators applied PacBio’s Iso-Seq method to establish a comprehensive transcript isoform resource for breast cancer, and identified thousands of breast tumor–specific splicing events, including 35 that are significantly associated with patient survival (of these, 21 are absent from GENCODE and 10 are enriched in specific breast cancer subtypes).
Read more about the HiFi Difference
More precise genomes for precision medicine
Not all gigabases are created equal