We’d like to extend a sincere thanks to everyone who attended our two-day North America User Group Meeting, held this year at our Certified Service Provider, the University of Delaware Sequencing and Genotyping Center (@UD_DNAcore). With representation from 80+ organizations and over 160 attendees, the event was a great environment for sharing best practices and networking with the SMRT Sequencing community. Also, a big thanks to our host, Bruce Kingham (@bkingham) and team, as well as our partners: Agilent, Biosoft Integrators, Circulomics, Covaris, Diagenode, Perkin Elmer, Sage Science and Shoreline Biome. If you weren’t able to attend the meeting, we’ve summed up the highlights below and you can download several of the presentations and view the recordings.


HiFi sequencing was a central theme for a few talks. Mitchell Vollger (@mrvollger) from the University of Washington spoke about using this approach to study segmental duplications in the human genome. The technique significantly reduced the complexity of accurately mapping these nearly identical sequences throughout the genome; it also reduced the amount of compute power needed compared to a previous PacBio assembly using continuous long reads instead of HiFi reads. Despite generating less data with the HiFi assembly, the team still resolved 30% more segmental duplications with the new approach.
PacBio scientist Meredith Ashby (@AshbyMere) also spoke about the benefits of HiFi reads, focusing her talk on metagenomics and microbiome characterization. She presented several examples of analysis — from full-length 16S sequencing to shotgun sequencing — showing how SMRT Sequencing enables accurate representation of these complex communities, in some cases even without fully assembling genomes. New updates will provide users with a dedicated microbial assembly pipeline, optimized for all classes of bacteria, as well as increased multiplexing on the Sequel II System, now with 48 validated barcoded adapters. That throughput could reduce the cost of microbial analysis to just $70 per sample, she noted.
While we’re on the subject of microbial sequencing, two lightning talks offered nice illustrations of this as well. Masako Nakanishi from the University of Connecticut Health Center presented a study of how the gut microbiome alters an organism’s susceptibility to colonic ulceration; next, she plans to examine cause and effect by evaluating results of fecal transplants in mice. Shawn Polson from the University of Delaware spoke about viral metagenomes, which are more challenging to distinguish than their bacterial counterparts because viruses have no 16S equivalent. With SMRT Sequencing, his team has generated higher-resolution data about viral genomes and aims to use this information as a guide to how these genomes function.
Iso-Seq Analysis
We also saw great presentations with varied applications of Iso-Seq data. Shawn Trojahn (@trojahn_shawn) from Washington State University presented results from transcriptome sequencing of grizzly bears. The analysis focused on differential gene expression during hibernation and active cycles, potentially offering human-relevant information about muscle atrophy and insulin resistance. Thanks to SMRT Sequencing, the team was able to identify more unique isoforms just from liver tissue than had been previously characterized in the entire reference genome. Of particular interest: more than 2,000 transcripts differentially expressed between hibernation and active season, including 86 genes that have isoforms expressed in opposite directions.
From the University of Wisconsin-Madison, Nic Wheeler (@wheeler_worm) spoke about RNA sequencing for filarial nematodes associated with understudied tropical diseases. His team used Iso-Seq analysis to improve gene models and achieve better transcriptome coverage for these worms, which typically have poorly annotated and fragmented genome assemblies. While getting enough RNA to study is a technical challenge, the group still managed to generate full-length isoforms, many of which were novel or contained novel junctions.
Ana Conesa (@anaconesa) from the University of Florida spoke about Iso-Seq analysis tools developed by her group, which created the popular SQANTI tools for Iso-Seq data QC. They’re also working on IsoAnnot to perform functional annotation at isoform resolution; validation has already been done on various species. Currently it’s a set of scripts, but her team is working to produce a more user-friendly version. Finally, tappAS is for functional diversity analysis and for prioritizing genes for validation.
We also had two Iso-Seq-focused lightning talks. Vince Magrini from Nationwide Children’s Hospital spoke about using Iso-Seq analysis as part of a comprehensive profiling strategy for pediatric cancer research; in one example, full-length isoform sequencing provided a clear view of a challenging mutation associated with a drug-targetable pathway. Alexandra Pike (@amimspike) at MIT presented a study of TIN2, a telomere-binding protein, which is mutated in some short telomere syndromes. By pairing the Iso-Seq method with CRISPR, her team revealed a previously uncharacterized TIN2 isoform that may have a functional difference for individuals with these syndromes.
Finally, PacBio scientist Kristin Mars spoke about recent updates, such as the single-day library prep that’s now possible with the Iso-Seq Express workflow. She also noted that one SMRT Cell 8M is sufficient for Iso-Seq experiments; that means whole transcriptome sequencing is feasible for $1,300 per sample, while multiplexed, targeted Iso-Seq analyses can cost as little as $185 per tissue.
Clinical Research
Two speakers offered attendees a clear view into potential future clinical use of SMRT Sequencing technology by showing how it’s performing in clinically oriented research labs. Melissa Smith (@lissagoingviral) from the Icahn School of Medicine at Mount Sinai shared results from using more than 1,300 SMRT Cells over the years — most of them for disease-focused research, but also covering microbial sequencing, immune profiling, epigenetics, ecology, and more. Her team has been working with the Sequel II System since January for applications ranging from honing targeted assays for disease-associated genes to performing targeted Iso-Seq for phasing drug targets with severity loci.
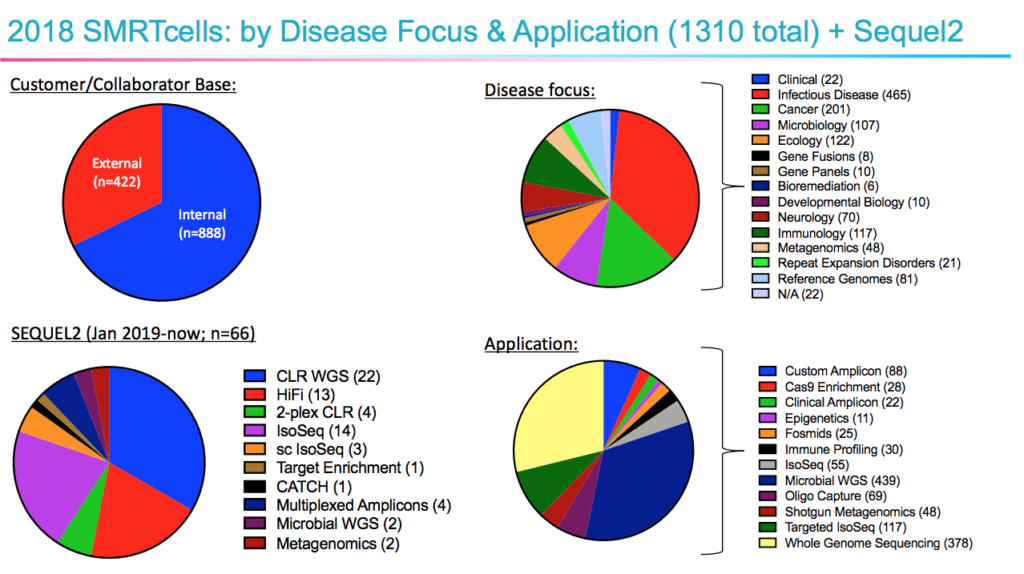
In a separate talk, LabCorp’s Brian Krueger (@h2so4hurts) discussed the use of SMRT Sequencing for clinical research related to HLA typing, viral genome sequencing, high-throughput variant confirmations to reduce the need for Sanger sequencing, and more.
Sample Preparation
We also had several speakers focused on technology development or sample prep protocols. Erin Bernberg (@ErinBernberg) from the University of Delaware reported on using the Agilent Femto Pulse for high-resolution, highly sensitive fragment analysis and on the low-input protocol, which her team used for a recent study of ice worms.
Eugenio Daviso from Covaris talked about the use of adaptive focused acoustics for gentle cell lysis and extraction of high molecular weight DNA. Mount Sinai’s Ethan Ellis presented results from the HLS-CATCH method, which involves the use of the SageHLS instrument with CRISPR design methods to target and extract large genomic fragments for sequencing while avoiding pseudogenes and other confounding regions.
In a lightning talk, NEB’s Kelly Zatopek shared data from RADAR-seq, an amplification-free method for detecting and quantifying a wide variety of DNA damage types across a genome. Finally, Shana McDevitt from the California Institute for Quantitative Biosciences shared the core lab perspective as she discussed sample size, purity requirements, and extraction protocols for PacBio sequencing.
We enjoyed connecting with many of our users in Delaware and learning about their latest discoveries and ideas for future research. A huge thanks to our wonderful SMRT Sequencing community for an engaging and exciting meeting!
November 21, 2019 | Events + conferences