
The genetic blueprint of every living organism, from humans to microbes, is written in DNA. This blueprint is shaped by a remarkable phenomenon: genetic variation. These variations, or differences in the DNA sequence, are what make each of us unique, contribute to susceptibility to certain diseases, and influence our response to the environment. For scientists, understanding these genomic variants is critical, not just for health research but also for advancing fields like agriculture and environmental sustainability.
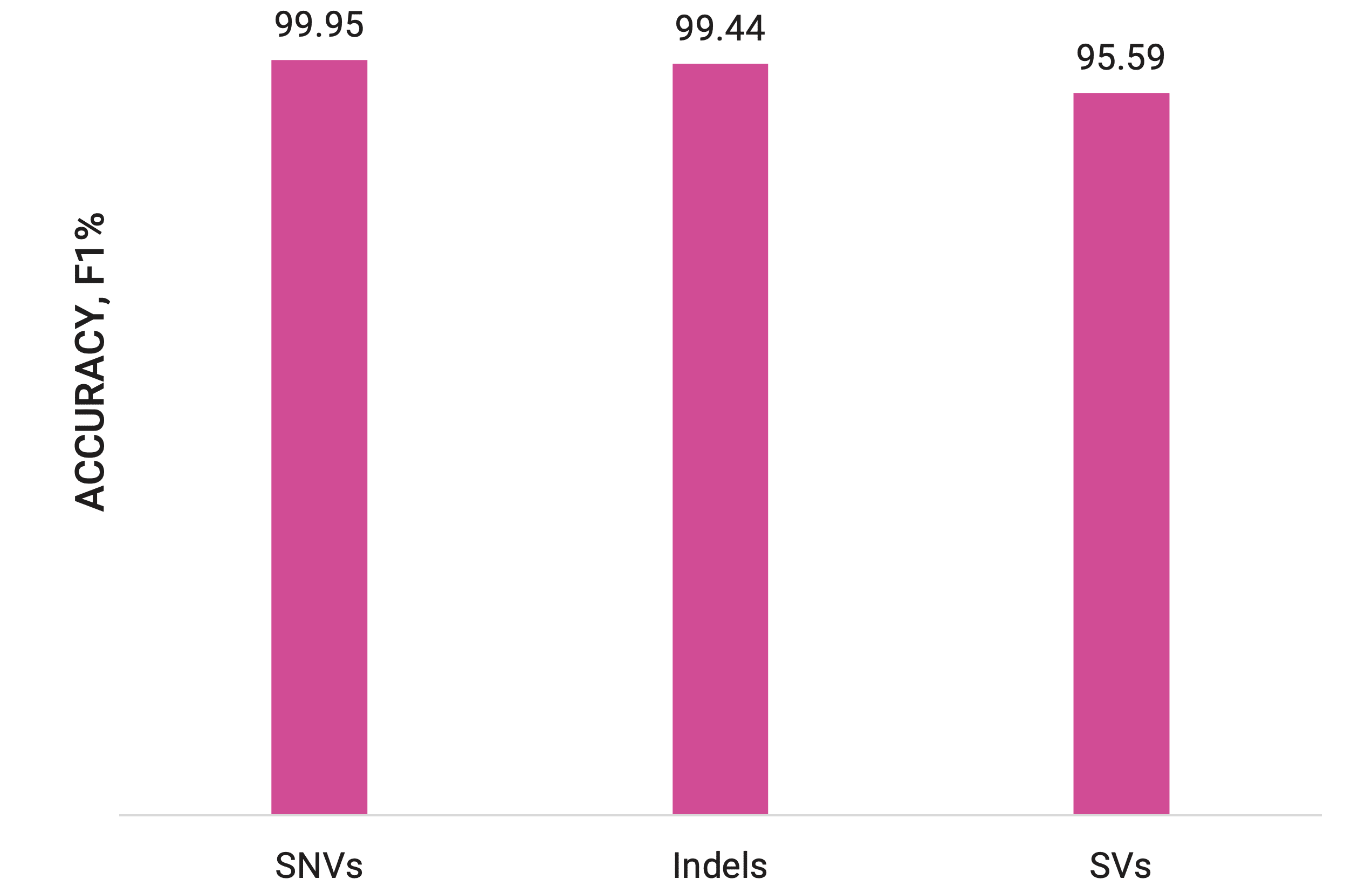
While the most common type of variant by count is the single nucleotide variant (SNV), other types—such as structural variants (SVs), indels (insertions and deletions), and copy number variants (CNVs)—can have far-reaching impacts on gene function and phenotype. PacBio sequencing solutions are uniquely equipped to help researchers detect all variant types with exceptional accuracy. Whether identifying variants linked to human disease or tracking changes in plant genomes, PacBio sequencing opens the door to a clearer, fuller picture of the genetic differences that shape life as we know it.

Single nucleotide variants (SNVs):
Small-scale changes with far-reaching impact
As the most prevalent type of genetic variation, single nucleotide variants (SNVs) occur at an extraordinary frequency. In every human genome, there are typically around 4 to 5 million SNVs. Each SNV represents a change at a single nucleotide—the basic building block of DNA. While a single base change might seem insignificant, the implications of SNVs are profound, especially when they occur in coding regions or regulatory elements of genes.
When SNVs occur within coding regions, they can result in amino acid substitutions, leading to altered protein structure and function. In some cases, this change can be benign, but in others, it can significantly impact biological processes and contribute to disease. Similarly, SNVs in regulatory elements can disrupt the normal control of gene expression, affecting when, where, and how much of a protein is produced. These disruptions have the potential to influence a wide array of conditions, including genetic diseases such as cystic fibrosis and sickle cell anemia, as well as complex diseases like cancer.
Indels:
Insertions and deletions that disrupt the code
Insertions and deletions (indels) represent another significant class of genetic variation, involving the addition or removal of small segments of DNA within the genome. Indels can range in size from just a single nucleotide to a few dozen base pairs, but even these seemingly minor changes can have dramatic effects on gene function. This is especially true when indels occur within coding regions of the genome, as they can disrupt the gene’s reading frame, a phenomenon known as a frameshift mutation. Even small indels can have a considerable impact on biological processes and are frequently associated with genetic diseases, like cystic fibrosis.
The detection of small variants like SNVs and indels requires a sequencing technology that prioritizes high accuracy to distinguish true variants from sequencing errors and can handle difficult-to-sequence regions to ensure comprehensive detection. The exceptional accuracy of PacBio HiFi sequencing provides high-resolution insights into these small genetic variants, making it easier to detect SNVs, even in challenging regions of the genome such as homopolymers or GC-rich sequences. This precision is equally critical for identifying indels, reducing the risk of missing critical mutations and providing a powerful tool for understanding genetic contributions to disease.
In addition to its strengths in detecting small variants, HiFi sequencing is also particularly well-suited for identifying larger genetic changes like structural variants, which involve more significant alterations in the genome.
Structural variants (SVs):
The giants of genetic variation
Structural variants (SVs) are larger genomic changes—typically greater than 50 base pairs—that can include large insertions, deletions, duplications, inversions, and translocations. While there are fewer SVs in the human genome compared to SNVs, they account for more genomic variation between individuals than SNVs and indels combined. The size and scope of SVs make them a significant source of genetic diversity, but also a major contributor to disease. For example, in cancer research, structural variants play a significant role in tumor progression by rearranging key genomic regions, resulting in the activation of oncogenes or the loss of tumor suppressor genes.
Detecting SVs has historically been a challenge due to their size and the complexity of the regions in which they often occur. However, HiFi sequencing offers exceptional clarity in detecting structural variants. The long read lengths of HiFi sequencing allow researchers to map large variants more accurately, even in repetitive or low-complexity regions of the genome. This capability is particularly valuable for understanding diseases such as autism, schizophrenia, and cancer, where SVs play a significant role in pathogenesis.
Copy number variants (CNVs):
Gene dosage matters
Copy number variants (CNVs) are a type of structural variant in which large sections of the genome are duplicated or deleted. These duplications or deletions can involve just one gene or large segments of chromosomes. CNVs are crucial in understanding diseases where gene dosage is important. For instance, CNVs have been linked to conditions like autism spectrum disorder, cancer, and developmental disorders.
Tandem repeats:
Repetitive but important
Tandem repeats are sequences of DNA in which a short segment is repeated multiple times in a row. While they are often seen as repetitive “junk” DNA, tandem repeats play an important role in gene regulation and genome stability. Variations in the number of repeats, known as repeat expansions or contractions, have been associated with diseases such as Huntington’s disease and Fragile X syndrome.
HiFi sequencing is particularly effective at resolving these complex variants due to its ability to capture intricate details of even the most difficult-to-sequence regions. HiFi read lengths enable the sequencing of repetitive and structurally variable regions, which are often challenging for short-read technologies. Once notoriously difficult to characterize, these regions can now be comprehensively genotyped at scale with the PacBio PureTarget repeat expansion panel for 20 of the most important repeat expansions for human health. HiFi technology now grants a clear view of these once-dark regions of the genome.
Epigenetic variants:
Beyond the DNA sequence
In addition to detecting genomic variants, HiFi sequencing can also simultaneously capture epigenetic changes, such as DNA methylation. Methylation is a chemical modification that occurs on DNA and plays a key role in regulating gene expression. Changes in methylation patterns are often associated with diseases such as cancer, where abnormal gene regulation can drive tumor development and progression.
With HiFi sequencing, researchers can detect both genetic and epigenetic variants in every read. This capability provides a more comprehensive view of how genetic and epigenetic changes interact with behavior and environment to influence phenotype, offering new insights into complex diseases.
Phasing variants for a more complete genetic picture
One of the key advantages of HiFi sequencing is its ability to phase variants, or determine which variants occur together on the same chromosome. This is important for understanding the full genetic context in which a variant occurs. For example, phasing can reveal whether two potentially harmful variants are located on the same chromosome or different ones, which can have important implications for disease risk and inheritance.
PacBio HiFi reads are well-suited for phasing because of their length and accuracy. Computational tools like HiPhase are used to phase both small and structural variants detected by HiFi sequencing. HiPhase benefits from highly accurate long reads that allow phase blocks to span breaks from reference gaps and homozygous deletions. Together this technology allows researchers to generate haplotype-resolved genomes, providing a more detailed understanding of how genetic variants are inherited and how they function together.
The role of variants in genetic research beyond human health
While human health research often dominates discussions about genomic variants, these changes in DNA also play an essential role in other areas of genetic research. In agriculture, for instance, genetic variants are key to breeding crops that are more resilient to environmental stressors, such as drought or pests. By identifying variants associated with these traits, scientists can develop crops that are better suited to changing climates, contributing to global food security.
Genetic variants can also influence how bacteria respond to pollutants or how they contribute to the health of ecosystems. Understanding these variations can help scientists develop strategies for environmental preservation and the management of natural resources.
The future of genomic research is powered by HiFi sequencing
As we look to the future of genomic research, the ability to accurately detect and understand genetic variants big and small will continue to be a driving force behind new discoveries. HiFi sequencing is at the forefront of this revolution, providing researchers with a more comprehensive view of the genome so that we can better understand how variants shape life as we know it and can use that knowledge to build a better future.
Explore the PacBio variant detection page to learn more about how HiFi sequencing powers the understanding of these variants and their impacts.