JULY 6, 2023 | SEQUENCING 101
シーケンシング入門:シーケンスカバレッジ
< Return to Japan blog | < Return to Main blog

この記事ではシーケンスカバレッジの基礎について説明します。高精度のロングリードシーケンスによって、どのカバレッジレベルでもゲノムの包括的な知見が得られるという重要な概念を発見してください。
シーケンスカバレッジとは?
ゲノミクスの専門家は、参照ゲノムまたはde novoアセンブリのある領域にアライメントするユニークなシーケンスリードの数を表すのに、「シーケンスカバレッジ」または「シーケンス深度」という用語を使用します。
30倍のヒトゲノムとは、リードがリファレンスの任意の領域に平均約30回アライメントすることを意味します。実用的には、シーケンス深度が高いほど、ゲノムのリード回数が多くなり、より正確で信頼性の高い情報が得られます。
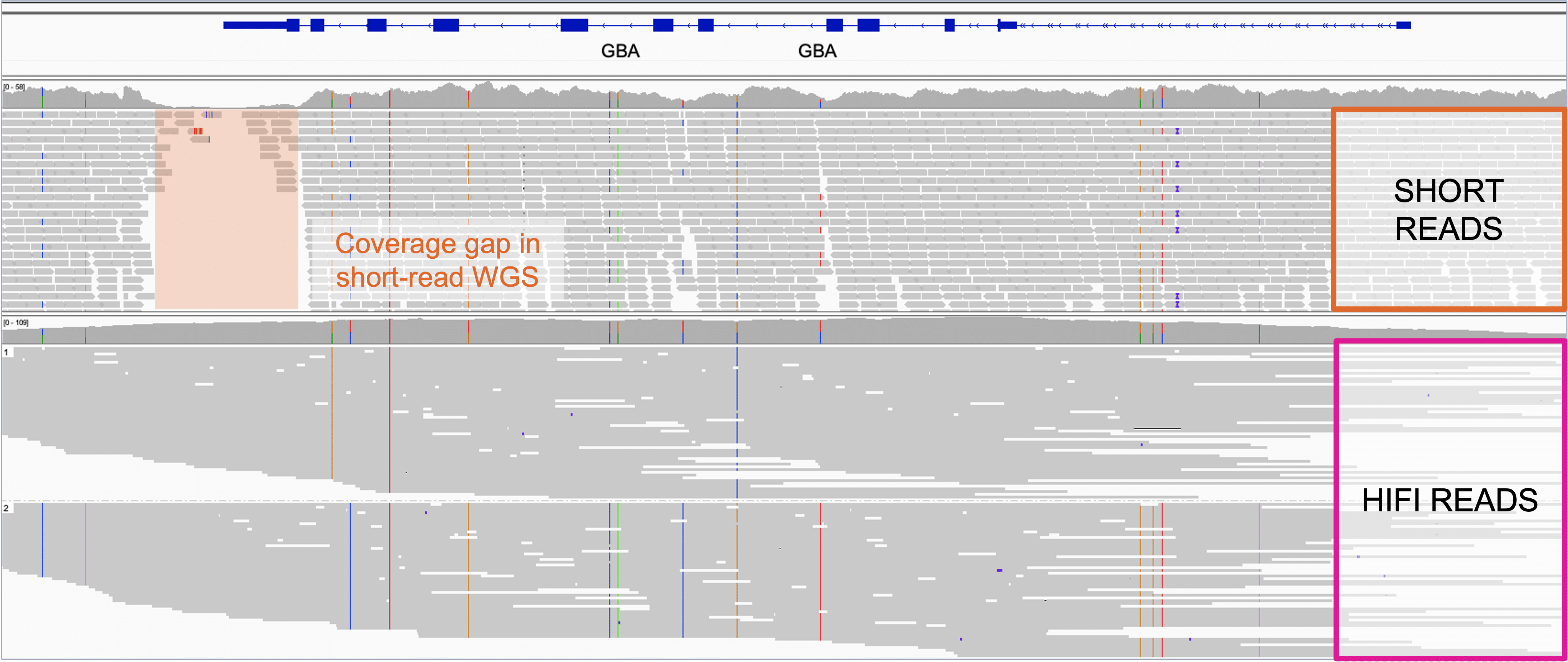 Figure 1. IGV generated image of PacBio long reads (purple section) and short-read alternative (orange section) covering a genomic reference region (blue line and bars at top). Note the area not covered by any reads (grey strips) in the short-read sequence alignment.
Figure 1. IGV generated image of PacBio long reads (purple section) and short-read alternative (orange section) covering a genomic reference region (blue line and bars at top). Note the area not covered by any reads (grey strips) in the short-read sequence alignment.
シーケンスカバレッジはなぜ重要なのですか?
シーケンスカバレッジがゲノム解析において重要なのは、カバレッジが高ければ高いほど、研究者の結果やそこから導かれる結論が正しいという統計的信頼性が高まるからです。
カバレッジを高めることは、科学者が、観察されたことが偶然やランダムなエラーではなく、生物学的サンプルの実際の属性であることを確信するために重要です。
科学において、実験結果に対する統計的信頼性を持つことは非常に重要です。コインを3回連続でひっくり返した場合、3回中2回は同じ側に出る可能性がある。もしそこで止めたとしたら、そのようなコインは片方の面に出ることが多いと結論づけるかもしれない(正確には66%の確率で)。しかし、3回というサンプルは少ない。もし、あなたが観察したコインの裏表が単なる偶然の結果だとしたらどうだろう?コインを30回ひっくり返したり、100回ひっくり返したりした場合、コインの表がどちらに出るかは50/50に近い結果になる可能性が高い。
シーケンスカバレッジが同じゲノムは科学的価値も同じか?
シーケンスカバレッジが等しいゲノムは、必ずしも科学的価値が等しいとは限りません。
ゲノムアライメントやアセンブリの説明力には多くの要因が影響します。しかし、ゲノムの生物学的価値に影響を与える基本的な要因(例:サンプルの質、実験デザインなど)に加えて、カバレッジの均一性と個々のリードの精度は、あるゲノムの科学的価値を他のゲノムよりも大きく高める可能性があります。この優れた例は、Saccharomyces cerevisiaeゲノムのde novoアセンブリーにおいて、高精度のロングリードPacBio HiFiデータによる20倍カバレッジが、ナノポアシーケンスによる20倍(実際には80倍の場合においても)カバレッジの有用性を上回ることを見出した、あるテクノロジー比較研究から得られたものです1。
カバレッジ均一性とは何か、なぜ重要なのか
カバレッジ均一性とは、個々のリードがゲノムまたは対象領域にわたってどれだけ均等に分布しているかを示すものです。
2つのゲノムを同程度のカバレッジ(例えば30倍)で配列決定しても、1つ目は均一性が低く(まったくカバーされていない遺伝子もあれば、60回カバーされている遺伝子もある)、2つ目はすべての遺伝子または領域が25~35回カバーされており、均一性が高い可能性があります。数字の上ではどちらも30倍ゲノムですが、前者は低品質で、ある領域ではギャップがあり、他の領域では優れたカバレッジがあります。一方、後者は全体を通して信頼性が高く、全ゲノムにわたる生物学の解釈に有用です。

どのくらいのシーケンスカバレッジが必要か?
研究の適切なシーケンスカバレッジレベルは、プロジェクトの目標や結果の適用方法によって大きく異なります。考慮すべき要素には以下が含まれますが、これらに限定されません:
使用するシーケンス技術の種類
- ゲノムの倍数性
- 研究したいバリアントや属性の複雑さや希少性
- サンプルの品質/劣化レベル
- 希望する統計的信頼度/検出力
- 査読付きジャーナルやデータリポジトリが定める要件
現在、ヒトゲノム解析では、30倍カバレッジが多くの生物医学研究分野におけるヒト全ゲノムシーケンス(WGS)の標準と広くみなされています。しかし、この基準はDNAシーケンスの化学的性質や能力が現在とは異なっていた時代に確立されたものです2。ショートリードsequencing-by-synthesis(SBS)技術の台頭以来、ゲノムの様々な側面を研究する際には、30倍を超えるカバレッジが必要であるとの指摘がなされています3,4。例えば、従来のSBSシーケンサーでは、ヒトのがん腫瘍を80倍以上のシーケンス深度で解析することがしばしば推奨されています5,6。ゲノム解析がショートリードNGSからロングリードシーケンスの時代に移行し始めると、私たちが探しているものを見つけるために必要なシーケンスカバレッジの量は、おそらくまた変化し始めるでしょう。

どのカバレッジレベルでも達成できることを再考する
科学へのアプローチ方法を再定義するような新技術が、時折登場します。全ゲノムシーケンスカバレッジの現在の標準は、ショートリードsequencing by synthesis(SBS)技術がまだ新しかった時代に確立されました2。その結果、30倍のシーケンスカバレッジベンチマークは、多くの点でその技術の能力と限界を反映しています。今日、DNAシーケンスの技術的状況はまったく異なっています。
新たな最前線は、高精度、高スループット、スケーラブルな技術に成熟したロングリードシーケンスによって定義されるようになりました。この新しいアプローチは、科学コミュニティに新境地を開くだけでなく、過去の研究、仮定、基準を再検討するよう求める形で、ゲノミクスの方法論的パラダイムを変えつつあります。
現在、PacBioロングリードWGSは、どのレベルのシーケンスカバレッジにおいても、SBSショートリードシーケンサーでは実質的に達成不可能な情報を提供することができます7。構造変異、ネイティブ5mCエピジェネティックコーリング、ハプロタイプ位相解析、そして、かつて「暗黒領域」と呼ばれていたもの(大きなリピート伸長、GCリッチ領域、セントロメア領域など)を含むゲノム全体の正確で均一なカバレッジは、現在では、Sequel IIeと新しいRevioシステムのどちらでも普段通りにPacBio HiFiロングリードシーケンスを実施すれば、データの一部として解析することが可能です。
ハイスループット・ロングリードシーケンスの時代において、ヒトゲノム解析におけるシーケンスカバレッジの新たな基準値はどうあるべきか、いまだ議論は続いています。とはいえ、旧来の標準が大きく変わろうとしていることは明らかであり、私たちはその行方をとても楽しみにしています。
PacBioのロングリードシーケンスが他のテクノロジーよりも必要カバレッジが低い理由をお知りになりたいですか?ロングリードシーケンスに関するこちらの記事をご覧ください。
低いカバレッジで豊富なゲノム情報が得られるのは素晴らしいことですが、そのコストは?
Revioシステムでは、10xヒトゲノムあたり約330米ドル*†で、真に「全体的な」全ゲノムを作成するためのゲノム情報の完全な補完的領域にこれまで以上に手頃な価格でアクセスすることができ、豊富な情報を得られるようになりました。このことは、ショートリードSBS技術で、相対的に少ない洞察を得るために必要な追加実験を省けることによって節約される時間、費用を考慮すると、特に真実だと言えます。30x PacBioロングリードヒトゲノムにご興味がある場合、Revioシステムは、995米ドルで、SMRTセル1つにつき1つの30x HiFiヒト全ゲノムを、バッチ処理なしで、~24時間のターンアラウンドで生産するように最適化されています。この30倍HiFi全ゲノムで、新規発見を可能にします。
これらの進歩はエキサイティングな可能性を提供しますが、1つの重大な疑問が生じます – あなたはどのような画期的な発見をするのでしょうか?
PacBioシステムへの投資、またはコアラボやサービスプロバイダーによるシーケンスにご興味はありますか?PacBioシーケンスを用いたプロジェクトの目標を達成するために、適切なカバレッジレベルを特定したいですか?
PacBioのサイエンティストにご相談ください。
References
-
Xue Zhang, Chen-Guang Liu, Shi-Hui Yang, Xia Wang, Feng-Wu Bai, Zhuo Wang, “Benchmarking of long-read sequencing, assemblers and polishers for yeast genome”, Briefings in Bioinformatics, Volume 23, Issue 3, May 2022, bbac146, https://doi.org/10.1093/bib/bbac146
-
Bentley, David R et al. “Accurate whole human genome sequencing using reversible terminator chemistry.” Nature 456,7218 (2008): 53-9. doi:10.1038/nature07517
-
Kong, Sek Won et al. “Measuring coverage and accuracy of whole-exome sequencing in clinical context.” Genetics in medicine: official journal of the American College of Medical Genetics 20,12 (2018): 1617-1626. doi:10.1038/gim.2018.51
-
Sims, D., Sudbery, I., Ilott, N. et al.Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15, 121–132 (2014). https://doi.org/10.1038/nrg3642
-
“Evaluating Somatic Variant Calling in Tumor/Normal Studies” https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/whitepaper_wgs_tn_somatic_variant_calling.pdf
-
Manja Meggendorfer et al. “Analytical demands to use whole-genome sequencing in precision oncology” Seminars in Cancer Biology, Vol 84, 2022, 16-22, https://doi.org/10.1016/j.semcancer.2021.06.009.
-
De Coster, W., Weissensteiner, M.H. & Sedlazeck, F.J. Towards population-scale long-read sequencing. Nat Rev Genet 22, 572–587 (2021). https://doi.org/10.1038/s41576-021-00367-3
-
William T. Harvey et al. “Whole-genome long-read sequencing downsampling and its effect on variant calling precision and recall” Preprint bioRxiv 2023.05.04.539448; doi: https://doi.org/10.1101/2023.05.04.539448
* 試験デザイン、サンプルタイプ、マルチプレックスのレベルは、必要な SMRT Cell の数に影響する可能性があります。費用は地域によって異なる場合があります。 価格には、Revioシステムで実行されるライブラリーとシーケンス試薬が含まれ、装置の償却やその他の試薬は含まれません。価格情報は2023年5月5日現在のものです。
†概算価格を達成するには、SMRTセル25Mあたりヒトゲノムライブラリーを3つマルチプレキシングする必要があります。
PacBioに問い合わせる