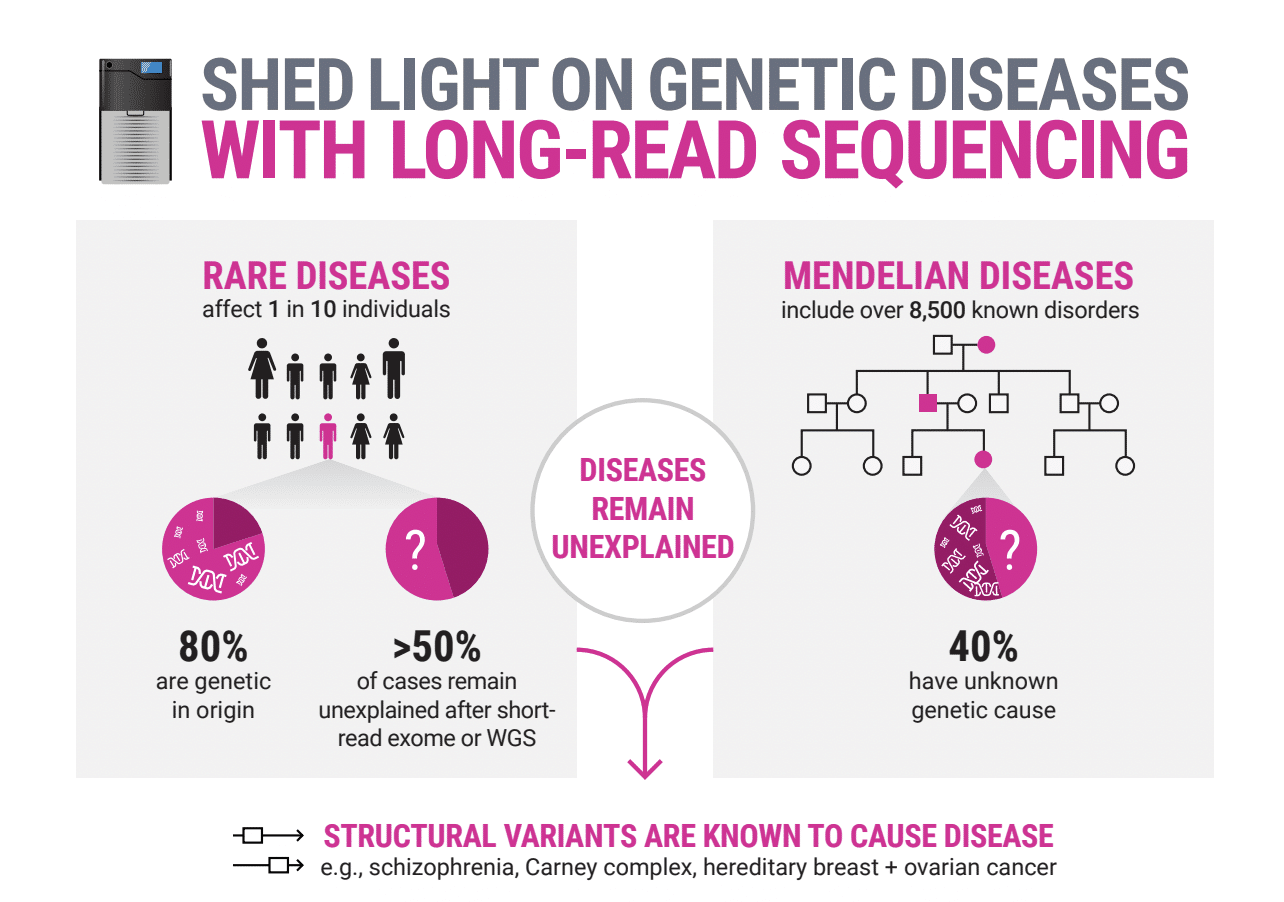
Rare diseases need better solutions
Although rare diseases are individually rare, collectively they are common, affecting 1 out of 15 people worldwide with an estimated >70% of cases being genetic in origin and >50% of cases remaining unsolved. This is despite many people undergoing diagnostic tests using approaches including microarrays, whole exome, or short-read whole genome sequencing.
Highly accurate long-read sequencing, known as HiFi sequencing, provides a higher resolution approach compared to previously used technologies to better understand the genetic causes of rare disease.
Whole genome sequencing
Generate more complete and phased human genome assemblies to better understand rare disease.
RNA sequencing
Help elucidate the functional impact of variants of unknown significance with full-length isoform sequencing using Kinnex.
Genetic testing
Efficiently capture clinically-relevant genes and variants on a single platform, while shedding light on complex regions of the genome.
HiFiSolves
Learn more about global initiatives that leverage long-read sequencing at scale to power rare disease research.
Whitepaper
Improving solve rates in rare disease with HiFi long-read sequencing
PacBio long-read sequencing offers significant advantages for rare disease research by allowing for highly accurate, comprehensive coverage of the genome. HiFi sequencing can capture large and challenging genomic regions and resolve complex structural variants (SVs), repetitive sequences, and segmental duplications, as well as provide phasing information that short reads often miss.

Spotlight
Rare disease research: My Scientific Journey
Hear from Dr. Vorasuk Shotelersuk, MD, FABMG (Chulalongkorn University, Bangkok, Thailand) on his research in rare disease and how he leverages HiFi sequencing to help identify rare and novel pathogenic variants.
Blog
Leading the way with first-tier HiFi sequencing
Researchers at Children’s Mercy Kansas City and Bioscientia are paving the way for long-read sequencing to replace legacy technology and reflex testing for clinical research, especially in rare disease.


Case study
Leveraging long-read whole genomes for rare disease research
Learn more about rare disease research and the advantages of long-read sequencing over legacy technologies. In this in-depth case study, discover how researchers at Children’s Mercy Kansas City use PacBio HiFi sequencing to help identify causative variants previously missed by conventional genetic testing, in order to provide answers for rare disease cases.
Spotlight
Understanding functional impact in rare disease
In this study, authors utilized full-length RNA sequencing with PacBio to elucidate the function of an intronic SNP thought to be causal in a case of axonal type 2a Charcot-Marie-Tooth disease. The study identified five distinct transcripts caused by alternative splicing, and found that while the canonical transcript was present, the candidate SNP led to deficient protein, a likely causal mechanism for the disease.

Spotlight
People first, because that’s where hope starts
Watch as Laura and Dave share their family’s rare disease journey, navigating home life with three young boys with an undiagnosed condition. The family is enrolled in a rare disease research study at Radboud University Medical Center (Radboudmc, Radboud, Netherlands) and hopes to gain further answers from researchers who are leveraging PacBio HiFi sequencing in the study.
On-demand webinar
Leveraging IsoForm-Level RNA Sequencing to Understand Rare Disease Pathogenesis
In a next-level exploration of the latest advancements in RNA sequencing technology and its application in the study of rare and inherited diseases, hear from Dr. Carolina Jaramillo Oquendo, a Research Fellow at the University of Southampton, as she presents her analyses of rare disease samples, in collaboration with the University Hospital Southampton NHS Foundation Trust.
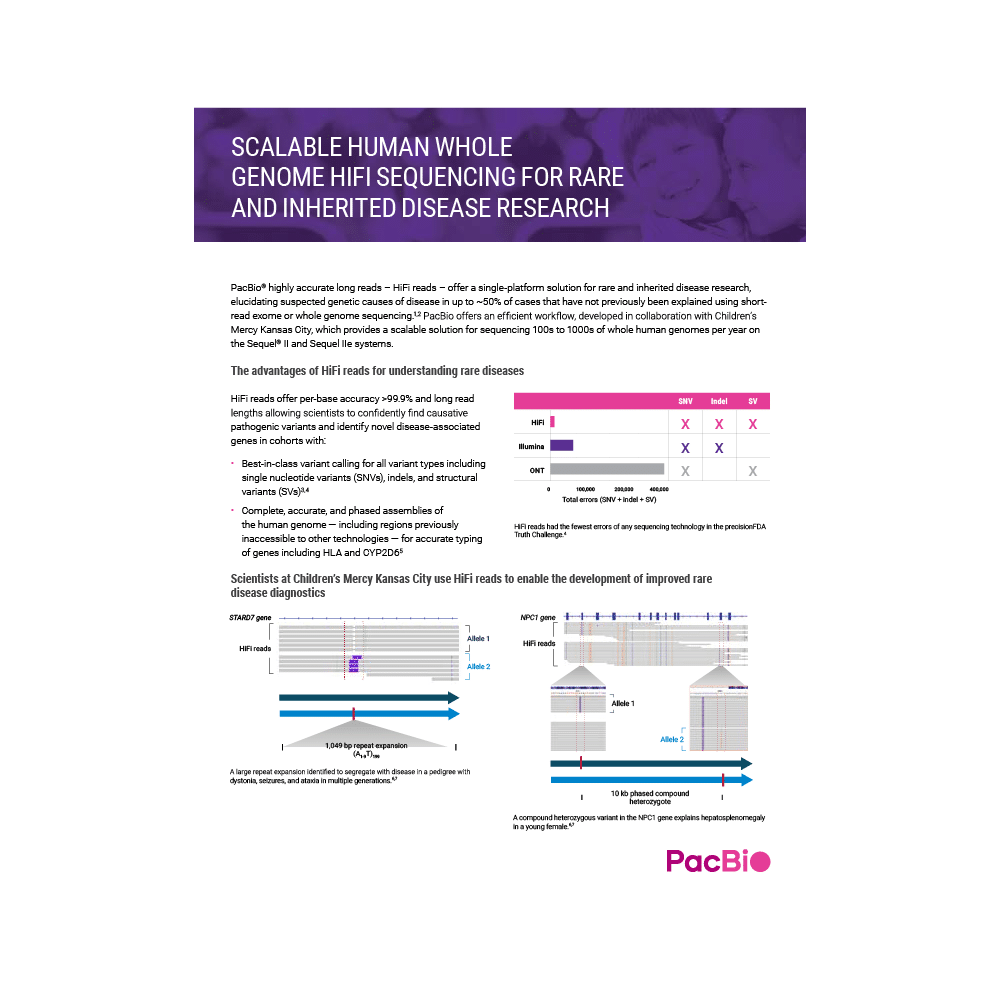
Scalable human whole genome HiFi sequencing for rare and inherited disease research
Explore our efficient and scalable workflow, developed in collaboration with Children’s Mercy Kansas City, for high-throughput sequencing and comprehensive variant detection to better understand the probable genetic causes of rare and inherited diseases.
Spotlight
A day in the life of a rare disease researcher
Learn more about the day-to-day work of Dr. Eliot Scherer, MD, PhD (Boston’s Childrens Hospital, Harvard Medical School) and how he leverages HiFi sequencing for his research to impact the lives of individuals and families with rare genetic hearing loss.
Spotlight
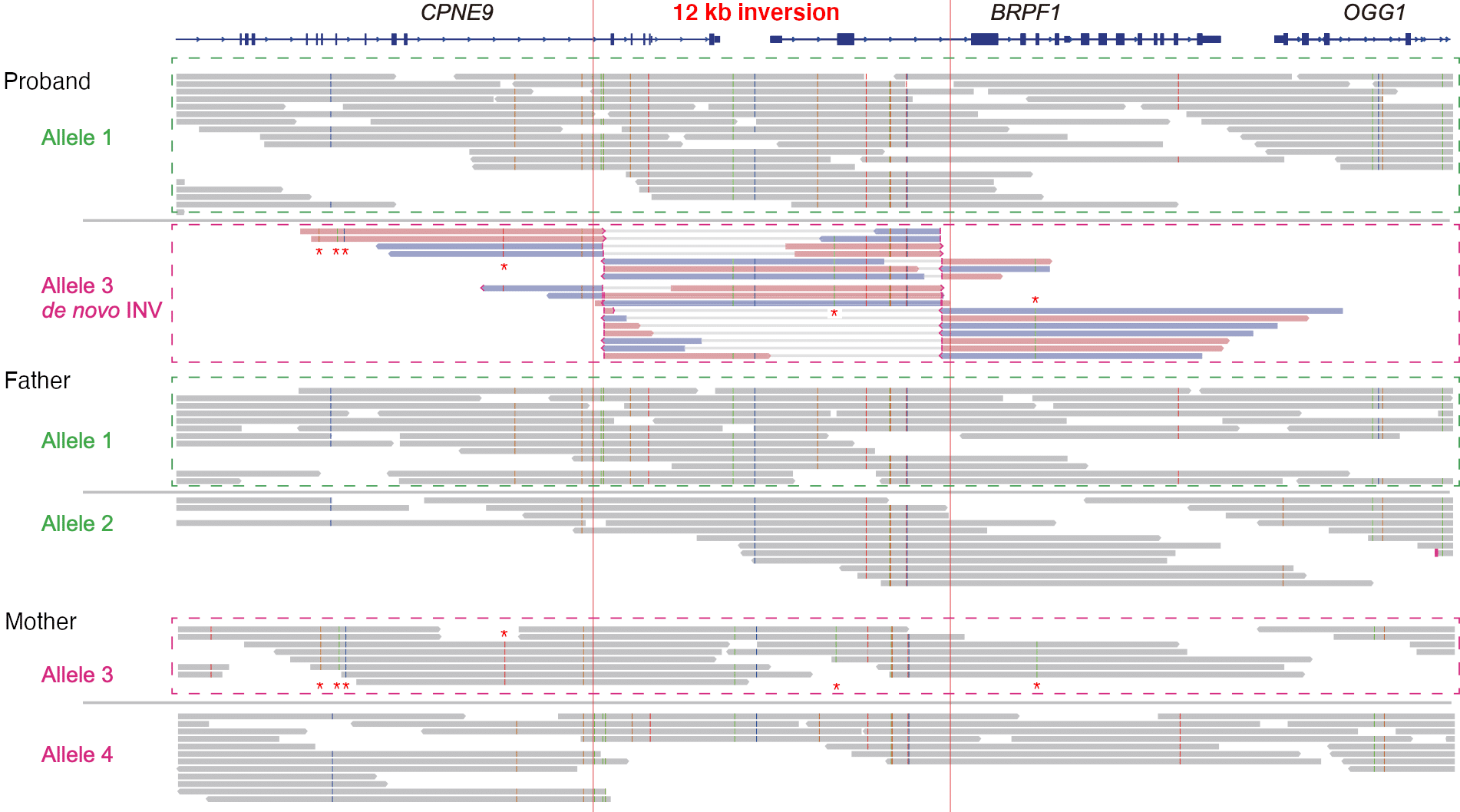
12 kb inversion identified as probable causative effect in syndromic intellectual disability
HiFi sequencing of a trio identifies a pathogenic heterozygous 12 kb de novo inversion that disrupts the BRPF1 gene. SNVs (marked with “*”) show that the inversion occurred on the maternal allele #3. Read more about this study in the blog post.
Mizuguchi, T., et al. (2021) Pathogenic 12-kb copy-neutral inversion in syndromic intellectual disability identified by high-fidelity long-read sequencing. d. 113(1).
Spotlight
Analyze and interpret your data with tertiary analysis partners
For RID research, tertiary analysis and interpretation is critical for getting the most value out of high-quality HiFi sequencing data. Learn more about PacBio WGS variant calling and explore our PacBio Compatible tertiary analysis partners.