Isoforms, not genes, are the drivers of disease and biology
In bulk RNA sequencing, short reads cannot span the entirely of transcripts and rely on computational assembly, which can often fail to resolve complex spliced isoforms. Now, you can perform full-length RNA sequencing with HiFi technology (Iso-Seq method) on the Vega system to see entire transcripts with no assembly required.

Highlights: (Jump to sections)
Key metrics

High-quality, full-length transcriptome – no assembly required
With the Kinnex full-length RNA kit and HiFi sequencing on the Vega benchtop system, you can obtain full-length transcript information up to 10 kb. Unambiguously characterize complex alternative splicing events and determine transcript start and end sites with confidence.
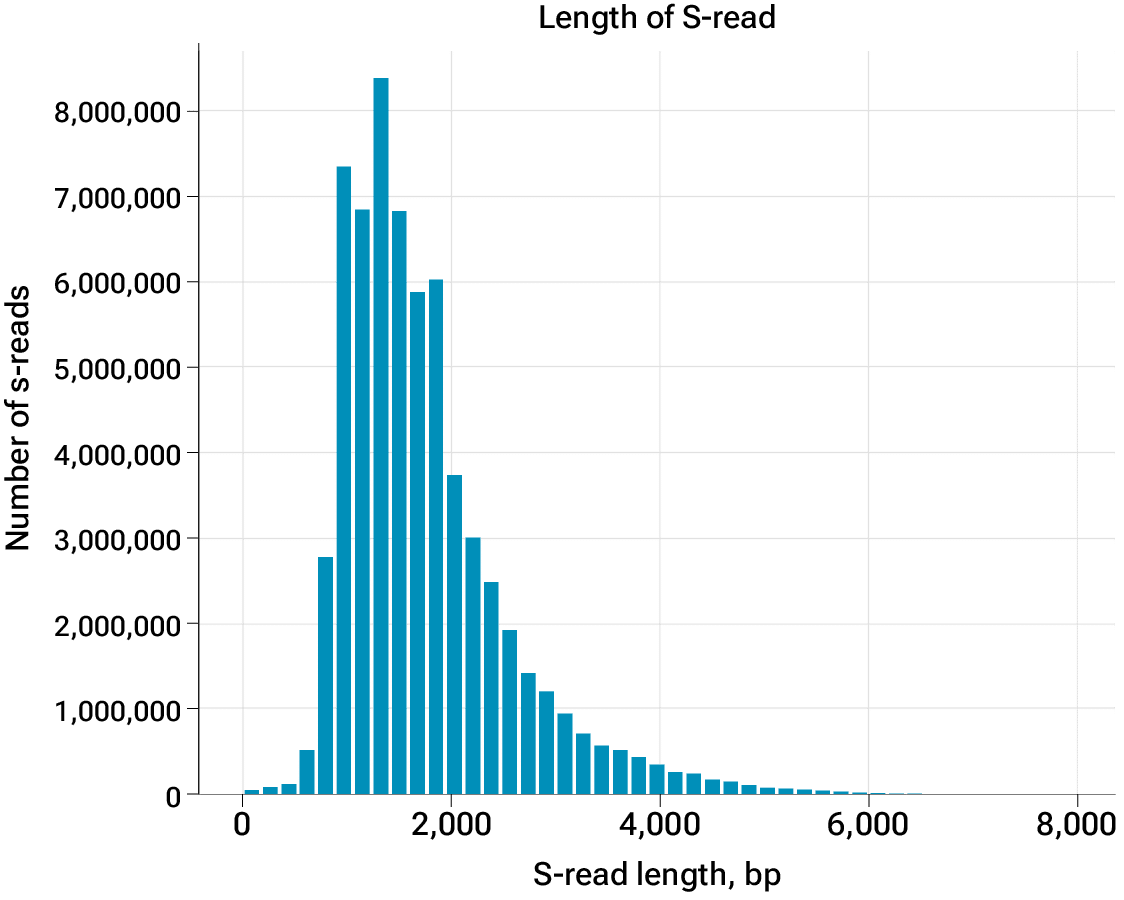
Figure 1. Transcript read length distribution for Kinnex full-length RNA data on the Vega system
Quantify and detect isoforms with allele specificity
Use SMRT Link combined with the latest informatics tools to accurately quantify isoform expression, detect fusion transcripts, predict open reading frames, and identify allele-specific isoform expressions.
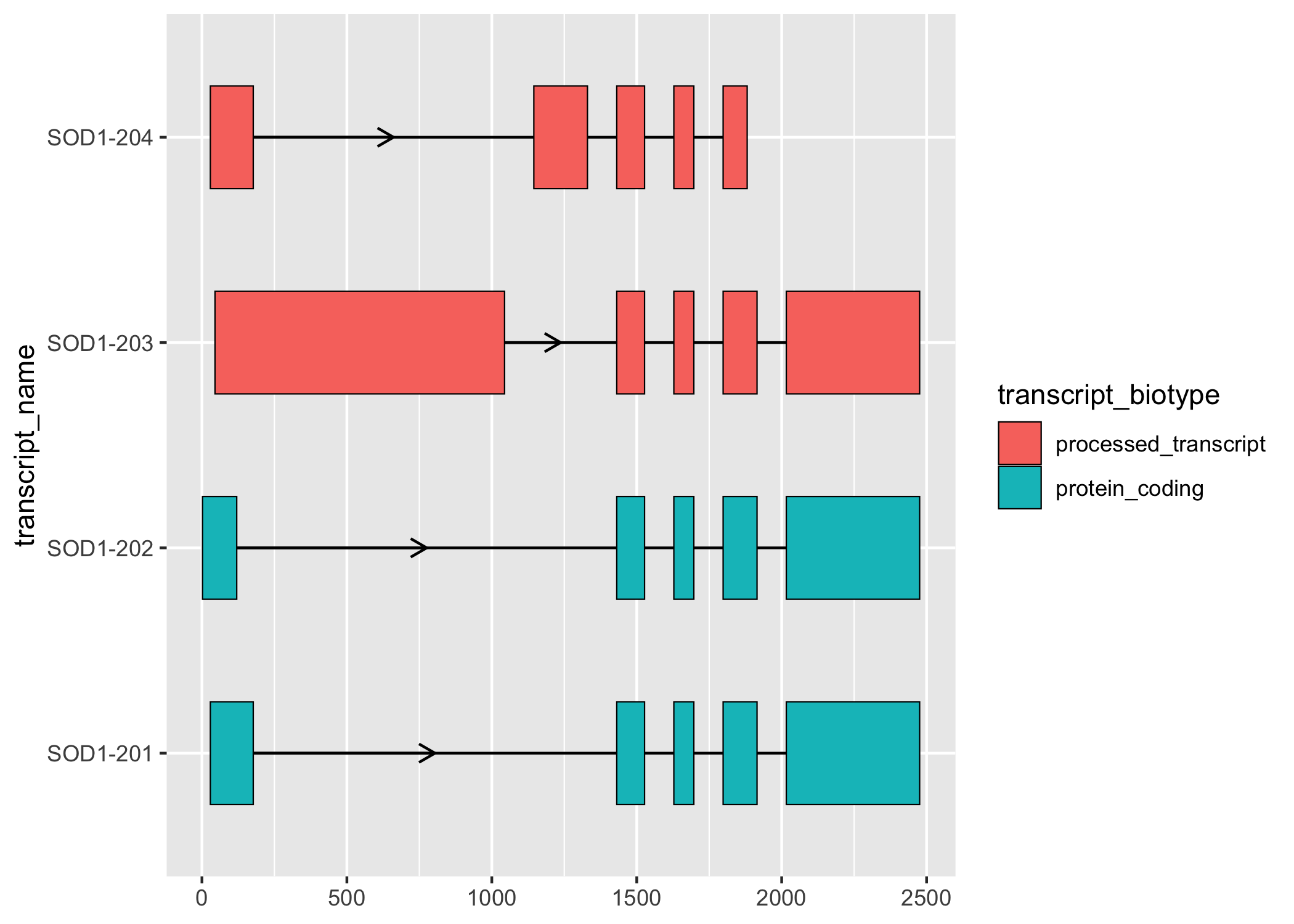
Figure 2. Kinnex full-length transcriptome data is compatible with many long-read RNA-Seq tools, including ggtranscript for visualization.
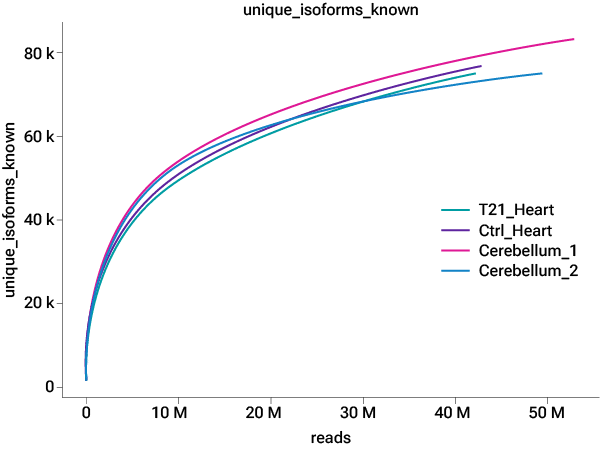
Figure 3. Saturation curve showing at 10 million reads, 80% of known genes & isoforms can be detected.
See more with less – multiplex for cost savings
With full-length reads that can span 1 to 10 kb, you can sequence with fewer reads to get the same high-quality information. For a standard quantification experiment, sequence 10—15 million reads per Vega SMRT Cell (2- or 3-plex). For larger cohort studies or highly expressed genes, sequence 5 million reads per Vega SMRT Cell (6-plex).
Kinnex full-length RNA sequencing workflow




Explore other Vega system datasets

Now you can
The Vega benchtop system makes high-accuracy sequencing affordable and accessible to labs of all sizes, bringing the power of HiFi sequencing within reach.